I
Вале́нтность (от лат. valentia - сила)
способность атома к образованию химических связей. Количественной мерой В. обычно принято считать число других атомов в молекуле, с которыми данный атом образует связи. В. - одно из фундаментальных понятий теории химического строения (см.
Химического строения теория). Оно формировалось вместе с понятием химической связи, параллельно с развитием синтетической химии и методов исследования строения и свойств веществ, и его содержание неоднократно расширялось и изменялось по мере того, как экспериментальная химия находила всё новые и новые классы соединений с неизвестными ранее типами взаимодействия атомов в молекуле, а в последние 30-40 лет - с развитием квантовой химии. В настоящее время накопленный химией экспериментальный материал столь обширен и разнообразен, а картина химической связи в разных соединениях столь пестра, что задача нахождения последовательного, единого и всеобъемлющего определения В. представляется крайне сложной. Эти трудности побуждают некоторых химиков вообще отказаться от поисков универсального понятия В. и заменить его набором более узких, но зато более конкретных и более точных понятий (ковалентность, гетеровалентность, координационное число и т.д.), область применимости каждого из которых ограничена соединениями с каким-либо одним преобладающим типом взаимодействия (ковалентным, ионным, координационным и т.д.). Однако до настоящего времени и в специальной, и в учебной литературе В. продолжает широко использоваться и как определение способности атома к образованию связей в самом общем смысле слова, и как количественная мера этой способности, и как синоним предлагаемых более узких понятий.
Для отдельных классов соединений, где преобладает какой-либо один тип химического взаимодействия, полезную информацию о способности атомов к образованию связей могут дать перечисленные ниже частные понятия (частные определения В.).
1. Определение понятия "валентность" и связь его с другими понятиями химии
Ковалентность - мера способности атома к образованию ковалентных химических связей, возникающих за счёт двух электронов (по одному от каждого атома) и имеющих малополярный характер (см. Ковалентная связь (См.
Купер)).
Ковалентность равна числу неспаренных электронов атома, участвующих в образовании связи, и часто может принимать все значения от 1 до максимальной, которая для большого числа элементов совпадает с номером их группы в периодической системе Менделеева (подробно см. разделы 2 и 3).
Гетеровалентность (употребляются также термины электровалентность и ионная
валентность) - мера способности атома к образованию ионных химических связей, возникающих за счёт электростатического взаимодействия ионов, которые образуются при полном (или почти полном) переходе электронов одного атома к другому (см.
Ионная связь). Гетеровалентность равна числу электронов, которые атом отдал или получил от другого атома, и совпадает с зарядом соответствующего иона (см. раздел 2).
Координационное число (КЧ) равно числу атомов, ионов или молекул, находящихся в непосредственной близости с данным атомом в молекуле, комплексном соединении или кристалле. В отличие от ковалентности и гетеровалентности, это понятие имеет чисто геометрический смысл и не зависит от характера связи между центральным атомом и лигандом. Так, например, КЧ атомов Al, Si, Р в комплексных ионах [AlFe6]3-, [SiFe6]2-, [PFe6]- равно 6, а КЧ атомов В, Xe, Ni в [ВН4]-, ХеО4, Ni (CO)4 равно 4. В кристалле NaCl каждый атом Na окружен шестью атомами Сl, так что КЧ Na равно 6. Величина КЧ может определяться как относительными размерами атомов, так и другими, более сложными причинами (см. разделы 2 и 3).
Окислительное число (ОЧ) (или степень окисления) - понятие, получившее в последнее время распространение в неорганической химии, - это электростатический заряд, условно приписываемый атому по следующим правилам. В ионных соединениях ОЧ совпадает с зарядом иона (например, в NaCl ОЧ Na равно +1, ОЧ Cl равно -1). В ковалентных соединениях ОЧ принято считать равным заряду, который получил бы атом, если бы все пары электронов, осуществляющие связь, были целиком перенесены к более электроотрицательным атомам (то есть если условно допустить, что связь имеет полностью ионный характер). Например, в HCl ОЧ Н равно +1, ОЧ Cl равно -1. В элементарных соединениях ОЧ равно 0 (например, в O2, Cl2, Р4, S8, в алмазе). При вычислении ОЧ в соединениях, где имеются два связанных атома одного элемента, их общую электронную пару принято делить пополам. Понятие ОЧ полезно при составлении уравнений окислительно-восстановительных реакций, для классификации неорганических и комплексных соединений и т.д.
Однако по своему определению ОЧ, в отличие от ковалентности и ионной В., имеющих чёткий физический смысл, носит в общем случае условный характер и, за исключением весьма ограниченного класса соединений с чисто ионной связью, не совпадает ни с эффективными зарядами атомов в соединениях, ни с фактическим количеством связей, которые атом образует. Кроме того, в ряде случаев, в частности, когда электроотрицательности двух разных связанных атомов близки и связь между ними имеет почти чисто ковалентный характер, возникает неопределённость, к какому из них следует целиком относить электронную пару (см.
Окислительное число).
2. Эволюция понятия "валентность" и его роль в истории химии
В начале 19 в. Дж.
Дальтоном был сформулирован закон кратных отношений, из которого следовало, что каждый атом одного элемента может соединяться с одним, двумя, тремя и т.д. атомами другого элемента (как, например, в окислах азота -N
2O, NO, N
2O
3, NO
2 и N
2O
5). В середине 19 в., когда были определены точные относительные веса атомов (И. Я.
Берцелиус и др.), стало ясно, что наибольшее число атомов, с которыми может соединяться данный атом, не превышает определённой величины, зависящей от его природы. Например, атом F может соединяться лишь с одним атомом Н, О - с двумя, N - с тремя, С - с четырьмя, образуя соответственно HF, H
2O, NH
3 и CH
4. Два или четыре атома Н в метане CH
4 могут быть замещены одним или двумя атомами О с образованием формальдегида CH
2O и двуокиси углерода CO
2 соответственно, три атома Н в CH
4 могут замещаться одним атомом N с образованием цианистого водорода HCN, и т.д. Эта способность связывать или замещать определённое число других атомов и была названа "В." (Э.
Франкленд, 1853).
В таком определении В., естественно, всегда выражается целыми числами. Поскольку в то время для водорода не были известны соединения, где он был бы связан более чем с одним атомом любого другого элемента, атом Н был выбран в качестве стандарта, обладающего В., равной 1. В "водородной" шкале кислород и сера имеют В., равную 2, азот и фосфор 3, углерод и кремний 4. Однако "водородной" шкалы оказалось недостаточно: в других соединениях, например в окислах, один и тот же элемент может реализовать В., которые не осуществляются в гидридах (существуют окислы P2O5, SO3 и Cl2O7, но неизвестны гидриды PH5, SH6 и ClH7). В качестве второго стандарта с В., равной 2, был выбран кислород.
В конце 50-х гг. 19 в. А. С.
Купер и А.
Кекуле постулировали принцип постоянной четырёхвалентности углерода в органических соединениях. Представления о В. составили важную часть теории химического строения А. М.
Бутлерова (1861) (см.
Химического строения теория). Образование химической связи рассматривалось как результат взаимного насыщения двух В. пары взаимодействующих атомов (по одной В. от каждого),
Кратные связи соответствовали насыщению нескольких В. от каждого атома, и т.д. Каждая связь считалась локализованной между двумя атомами и изображалась одной чертой, соединяющей эти атомы. Молекулы стали изображать с помощью структурных формул, получивших особенно широкое распространение в органической химии.
Положения Бутлерова в дальнейшем легли в основу структурной теории, рассматривающей и пространственное расположение атомов в молекуле. Было найдено, что простые молекулы типа MXk с одинаковым центральным атомом M и разными заместителями Х имеют схожее геометрическое строение. Независимость геометрического строения от типа связи в широких пределах привела к мысли, что пространственное расположение атомов в молекулах MXk определяется В. центрального атома М и что эти В. имеют направленный характер (см. раздел 3).
Периодический закон Д. И. Менделеева (1869) вскрыл зависимость В. элемента от его положения в периодической системе (см.
Периодическая система элементов Д. И. Менделеева). Элементы одинаковых групп системы обладают одинаковой высшей В., в большинстве случаев равной номеру той группы, в которой находится этот элемент; высшая В. меняется на 1 при переходе от одной группы к соседним. Эта зависимость сыграла чрезвычайно важную роль в развитии химии: зная лишь положение элемента (в том числе элементов, которые в то время ещё не были открыты) в периодической системе, можно было определить его валентные возможности, предсказать состав его соединений и впоследствии синтезировать их. С помощью представлений о формальной (стехиометрической) В. (см.
Стехиометрия) химикам удалось обобщить и систематизировать огромный экспериментальный материал по строению, стехиометрическому составу и свойствам многих десятков и сотен тысяч органических и неорганических соединений.
Первые электронные теории ковалентности и гетеровалентности. До электронных представлений о строении вещества В. трактовалась формально. Лишь в 20 в. было установлено, что химическая связь осуществляется за счёт электронов внешних (валентных) оболочек атомов.
В 1916 Г.
Льюис постулировал, что химическая связь осуществляется парой электронов, принадлежащих одновременно обоим взаимодействующим атомам. В 1917 В.
Коссель выдвинул гипотезу, согласно которой электронная пара связи переходит целиком к одному из атомов с образованием ионной пары катион - анион, удерживающихся в молекуле электростатическими силами. Согласно обеим гипотезам наиболее устойчивыми оказываются соединения, в которых валентные электроны распределялись так, чтобы каждый атом был окружен оболочкой, имитирующей электронную оболочку ближайшего инертного газа (правило октета). Гипотеза Льюиса положила начало электронной теории ковалентной связи и ковалентности, гипотеза Косселя - теории ионной связи и гетеровалентности. Обе представляли крайние случаи общей картины полярной связи, когда электронная пара смещена к одному из атомов лишь частично и степень смещения может варьировать от 0 до 1 (см.
Полярность химических связей). В. атома в соединении, согласно классической электронной теории, равна числу его неспаренных электронов, участвующих в связях, а максимальная В. - обычно полному числу электронов в его валентной оболочке, то есть номеру труппы периодической системы, в которой находится элемент. Элементы одинаковых групп имеют одинаковое число валентных электронов, а внутри одинаковых подгрупп - и одинаковые или очень близкие электронные конфигурации (см. раздел 3). Сходство строения валентных оболочек атомов обусловливает сходство их соединений.
Ковалентность и гетеровалентность отражают специфику соответствующего типа химической связи. Для ковалентности важна насыщаемость связей, обусловливающая существование молекул в виде дискретных частиц с определённым составом и структурой. Ковалентность эффективна для органических и большинства простых неорганических соединений. Напротив, в случае гетеровалентности максимальное число ионов противоположного знака, способное разместиться вокруг данного иона, в основном определяется соотношениями их размеров. Ионная В. эффективна для сравнительно ограниченного класса соединений, в основном для различных солей щелочных, щёлочноземельных и некоторых др. металлов.
В. в комплексных соединениях. Ещё в конце прошлого века было найдено (А.
Вернер, 1893), что многие соединения, как с максимальными (насыщенновалентные), так и с промежуточными В., типа ВСl
3, SiCl
4, PCl
5, CrCl
3 и т.п., обладают склонностью к взаимодействию с другими насыщенновалентными соединениями - солями, окислами, молекулами типа H
2O, NH
3 и др., с образованием довольно прочных комплексных соединений - K [BCl
4], K
2[SiCl
6], NH
4[PCl
6] и т.д. Исследования их строения рентгеновскими методами показали, что в комплексных анионах
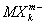
и катионах

атомы лигандов Х обычно находятся в вершинах правильных многоугольников (октаэдра, тетраэдра и др.), а все связи М - Х одинаковы.
Для представлений о В. комплексные соединения необычны тем, что в них координационное число КЧ может быть больше общего числа валентных электронов атома М. Более того, в парамагнитных (см.
Магнетохимия) комплексах переходных и редкоземельных металлов - K
4[CrF
6], K
3[CrF
6], K
2[CrF
6] и др., некоторые электроны валентной оболочки остаются неспаренными и локализованными у центрального атома и практически не участвуют в связи. Классическая В. и КЧ, как правило, не совпадают, а способность к образованию октаэдрических и тетраэдрических. комплексов оказалась чрезвычайно распространённой и типичной для многих металлов и неметаллов, связанной сложной зависимостью с положением элемента в периодической системе и его В. в исходном простом соединении.
Поэтому было высказано предположение, что, наряду с "классической" В., которая реализуется в исходных простых соединениях типа ВСl
3, SiCl
4 и др., атомы обладают также "координационной" В. (см.
Донорно-акцепторная связь), которая насыщается в комплексных соединениях (о природе координационной В. см. раздел 3). Предпринимались попытки описать связь в комплексных соединениях в рамках ионной теории, в которой считается, что анионы типа [PF
6]
- и [МnO
4]
- построены из ионов P
5+ + 6F
- и Mn
7+ + 4O
2- и что В. центрального атома совпадает с зарядом его иона. Однако затраты энергии, необходимой для перевода 1 атома Mn и 4 атомов О в состояния Mn
7+ и O
2-, далеко не компенсируются выигрышем в энергии при образовании связи. С появлением экспериментальных методов определения эффективных зарядов стало ясно, что эффективные заряды вообще редко превышают значения +1 или +2 у положительно заряженных и -1 у отрицательно заряженных атомов и обычно выражаются дробными долями заряда электрона (в перманганатном анионе заряд на Mn составляет лишь +1,5-+2,0 электрона). Поэтому ионная теория для большинства неорганических соединений, простых и комплексных, не может считаться корректной.
Успехи химии 20 в. и проблемы теории В. В 20 в. экспериментальной химией было синтезировано и изучено строение множества новых соединений, которые также оказалось невозможно уместить в рамки классических представлений о В. Оказалось, что склонность к образованию координационных соединений и насыщению координационных В. вообще чрезвычайно распространена и характерна практически для всех элементов и что суждения о В. на основании одного лишь стехиометрического состава очень часто оказываются несостоятельными без точных данных о структуре соединения и геометрическом расположении ближайшего окружения рассматриваемого атома. По мере развития структурных методов (см.
Электронография молекул,
Рентгенография молекул) стало известно, что многие соединения с простым брутто-составом (AlCl
3, PdCl
2, MoO
3 и др.), ранее считавшиеся простыми, в действительности даже в пара́х имеют димерное и полимерное строение - Al
2Cl
6, (PdCl
2)
x (
рис. 1, а, б), (MoO
3)
2-5. В них "мостиковые" лиганды, соединённые одинаковыми связями с двумя атомами металлов (на рис. 1 они помечены цифрой 2), обладают координационным числом КЧ = 2. У соединений в твёрдом состоянии, которые часто построены ещё сложнее, КЧ галогенов и кислорода, ранее выбранного в качестве стандартного двухвалентного элемента, могут быть 3 и даже 4. В бороводородах (См.
Бороводороды) каждый "мостиковый" атом водорода, считавшегося ранее стандартным одновалентным элементом, связан одинаковыми связями с двумя атомами бора (
рис. 1, б). Алкильные группы также способны образовывать мостиковые связи в металлоорганических соединениях (См.
Металлоорганические соединения) типа Al
2(CH
3)
6 (
рис. 1, г) и др.
Для соединений переходных и ряда непереходных элементов оказалось характерным использование дополнительной В. за счёт образования связей металл - металл (кластерные соединения), при этом расстояние между атомами металлов оказалось значительно меньшим, чем в индивидуальных металлах. Например, в дигалогенидах молибдена и вольфрама во многих химических реакциях сохраняется неизменной группа
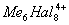
(
рис. 2), в которой атомы металла (Me) образуют правильный октаэдр; каждый атом Me связан с четырьмя другими атомами Me и с четырьмя атомами галогена (Hal), а каждый атом Hal связан с тремя атомами Me. Связи Me - Me в кластерах могут быть кратными (как, например, в
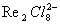
, где расстояние Re - Re на 0,5 Å меньше, чем в металлическом Re, и на их образование атомы могут тратить не одну, а несколько В.
Недостаточность классического понимания В. видна также на примере так называемых "нуль-валентных" соединений, где атом металла связан исключительно с нейтральными молекулами; таковы
Карбонилы металлов типа Ti (CO)
7, Cr (CO)
6, Fe (CO)
5,
Аммиакаты типа Pt (NH
3)
4 и т.д. В них вообще отсутствует классическое валентное взаимодействие (у атомов С и N в молекулах CO и NH
3 нет неспаренных электронов), а связь осуществляется только за счёт координационных В. атома металла и молекул лигандов. Нейтральные лиганды часто оказываются мостиковыми и образуют по две, например в Co
4(CO)
12, и даже по три, например в Rh
6(CO)
16, связи.
Для теории В. особый интерес представляют так называемые π-комплексы переходных металлов с ароматическими молекулами или молекулами с сопряжёнными связями в качестве лигандов (этиленом, циклопентадиенилом, бензолом и др.) типа ферроцена Fe (C5H5)2, дибензолхрома Cr (C6H6)2 (рис. 3, а, б), тетрациклопентадиенила титана Ti (C5H5)4 и др. В отличие от комплексов типа [Сr (NHз)6]3+, [Сr (H2O)6]2+ или Cr (CO)6, где центральный атом осуществляет связь с лигандом через один атом от каждого лиганда (через N - в аммиакатах, через О - в гидратах, и т.д.), в π-комплексах атомы Fe, Cr и Ti взаимодействуют совершенно одинаково со всеми атомами С каждого ароматического кольца. Непригодность классической В. или КЧ здесь очевидна: при этом пришлось бы считать все атомы углерода 5-валентными, а атомы Fe, Cr и Ti - соответственно 10-, 12- и 20-валентными. Единственный неспаренный электрон, который имеется у радикала ․C5H5 (так же как и у многих других ароматических радикалов типа тропила ․C7H7 и т.д.), в равной степени принадлежит всем углеродным атомам кольца. Для этого класса соединений потребовались представления о делокализованной ("групповой") В., характеризующей всю совокупность атомов С в ароматическом кольце.
Сейчас стало ясно, что КЧ в комплексах, так же как В. в простых соединениях, не является жестко специфической характеристикой элемента: для большого числа металлов были найдены комплексы со всеми промежуточными значениями КЧ от 3 до 7, 8 и 9. При этом возникли трудности с самим определением КЧ: в низкосимметричных высококоординационных комплексах расстояния М - Х, даже для одинаковых лигандов X, часто оказываются неодинаковыми; при этом они могут быть больше тех достаточно коротких расстояний, при которых наличие сильного взаимодействия бесспорно, но всё же недостаточно велики, чтобы их можно было уверенно исключить из координационной сферы комплекса.
Новые проблемы В. возникли и в других разделах химии. Сильное развитие получила химия свободных радикалов [например, метил ․CH
3, трифенилметил
․C (C
6H
5)
3 и др., см.
Радикалы свободные], в которых имеются атомы 3-валентного углерода. В последнем десятилетии были синтезированы соединения инертных газов (См.
Инертные газы) типа XeF
2, XeF
4, XeF
6, XeO
3 и др., то есть соединения элементов, которые ранее считались вообще неспособными к химическому взаимодействию. Стало ясно и то, что В. элементов может сильно меняться с изменением внешних условий, в частности температуры. Например, PCl
5, существующий при умеренных температурах в газовой фазе в виде мономерных молекул, при конденсации диспропорционирует (см.
Диспропорционирования реакция),
давая пару катион [РСl
4]
+ (КЧ = 4) - анион [РСl
6]
- (КЧ = 6). Наоборот, при повышении температуры обнаруживаются молекулы PCl
3, PCl
2, PCl, ионы PCl
4+, PCl
3+, Pd
2+, PCl
+ и т.д. Благодаря успехам химии молекул в газовой фазе за последние 20 лет найдено огромное число соединений (часто сложного состава) с промежуточными и необычными В., которые не обнаруживаются у соединений в обычных условиях. Например, кроме давно известных анионов типа CO
32- и SO
42-, сейчас обнаружены анионы CO
3-, SO
4- и нейтральные молекулы CO
3, SO
4. Кроме насыщенных молекул типа CH
4, C
2H
6, найдены ионы типа CH
5+, C
2H
7+, кроме молекулы H
2 - ион Н
з+, и т.д.
Сейчас установлено, что подавляющее большинство элементов может проявлять переменную В., образуя весь ряд "валентноненасыщенных" соединений со всеми значениями В. от 1 до максимальной с изменением на 1 (например, известны молекулы BF, BF2 и BF3; CF, CF2, CF3 и CF4 и т.д.). В. не может считаться жестко специфической характеристикой элемента, можно говорить лишь об относительной типичности или относительной устойчивости разных значений В. У непереходных элементов чётных и нечётных групп наиболее устойчивы соответственно чётные и нечётные В., например в молекулах типа PF3, PF5, SF2, SF4, SF6, IF, IF3, IF5, IF7 и т.д., где типичная В. атомов Р, S и I изменяется на 2 единицы.
Радикалы типа ·PF4, ·SF3, ·SF5, ·IF2, ·IF4 и т.д. с четырёхвалентным фосфором, нечётновалентными аналогами серы и инертными газами и чётновалентными галогенами значительно менее стабильны, обладают отчётливо выраженной склонностью к отщеплению одного электрона (с образованием более устойчивых катионов типа PF4+, SF3+, SF5+, IF2+, IF4+) или одного атома заместителя и характеризуются значительно меньшими временами существования. У элементов побочных групп соотношения между типичными и менее типичными В. имеют более сложный характер.
Изучение электронных спектров показало, что двухатомные молекулы типа O2, S2, OS и др. имеют два неспаренных электрона; в рамках классических представлений это следовало бы интерпретировать так, будто в подобных молекулах каждый атом сохраняет неиспользованной одну свою В., хотя нет никаких видимых препятствий для их использования.
До сих пор не решена проблема В. в случае интерметаллических соединений (см.
Металлиды,
Металлическая связь), имеющих обычно сложный состав типа Cu
5Zn
8, Cu
31Sn
8, Zn
21Fe
5, нестехиометрических окислов, нитридов, карбидов, силицидов и других соединений металлов, в которых состав может меняться непрерывно в сравнительно широких пределах.
Таким образом, поиск общего определения В., охватывающего все известные типы соединений и тем более способного предсказать возможность или принципиальную невозможность существования ещё не известных классов соединений, представляет сложную проблему. Конечно, параллельно с "неклассическими" соединениями химиками были синтезированы многие сотни тысяч соединений, которые могут быть интерпретированы в рамках обычных классических представлений о В. Однако ясно, что все существующие частные определения В. (см. раздел 1) ограничены определёнными классами и типами соединений, в которых преобладает какой-либо один тип химического взаимодействия. В общем же случае связи имеют промежуточный характер между чисто ионными и чисто ковалентными, в них принимают участие все типы взаимодействия одновременно, но в различных количественных соотношениях, резко изменяющихся от класса к классу и более плавно - от соединения к соединению внутри одного класса. При отсутствии общего определения В. трудность заключается в том, чтобы определить границы, где перестаёт быть справедливым одно частное определение В. и его заменяет другое. Решить эту проблему только на основании экспериментальных фактов и классических представлений невозможно. Существенную помощь здесь может оказать квантовая теория химической связи и В.
3. Современные квантово-химические представления о валентности
Начиная с 30-х гг. 20 в. представления о природе и характере В. постоянно расширялись и углублялись, параллельно с расширением и углублением представлений о химической связи. Существенный прогресс был достигнут в 1927, когда В.
Гейтлер и Ф. Лондон выполнили первый количественный квантово-химический расчёт молекулы H
2. В подтверждение гипотезы Льюиса было показано, что химическая связь в H
2 действительно осуществляется парой электронов и является результатом электростатического (кулоновского) взаимодействия электронов и ядер. Образование молекулы из атомов энергетически выгодно, если
Спины электронов направлены в противоположные стороны, когда притяжение электронов к ядру (остову) чужих атомов больше энергии отталкивания между электронами и между ядрами. Параллельная ориентация спинов приводит к отталкиванию атомов друг от друга.
В дальнейшем идеи Гейтлера - Лондона были распространены на многоатомные молекулы, что привело к созданию теории локализованных пар. Согласно этой теории, общая картина распределения электронной плотности в молекулах типа MX
k складывается из независимых фрагментов М - X, связь в каждом из которых осуществлена парой электронов (по одному от центрального атома М и от заместителя X), локализованной между двумя атомами М и X. Согласно этой теории В. не просто связывается с наличием неспаренного электрона, но и характеризуется тем, в каком состоянии этот электрон находится (см.
Атом.) или, в терминах теории химической связи, какую атомную орбиталь (АО) он занимает. АО разного типа имеют различную ориентацию в пространстве: s-орбиталь сферически симметрична, орбитали
px,
ру и
pz вытянуты вдоль трёх взаимно перпендикулярных осей и т.д. Электроны атомов в молекулах в общем случае описываются "гибридными" (смешанными) орбиталями, в которые, в принципе, могут входить любые валентные АО в разных количественных соотношениях и у которых электронные облака сконцентрированы вдоль направлений связей М - Х значительно сильнее, чем у простых АО. Состояние валентных электронов, а следовательно и свойства В. атома М, в значительной мере определяют закономерности в свойствах молекул MX
k для широкого круга заместителей X. Наиболее плодотворными оказались концепции направленных В. и валентных состояний атомов, позволившие объяснить и обобщить ряд закономерностей в геометрическом строении и энергиях химических связей органических и неорганических молекул.
В теории направленных валентностей предполагается, что связи М - Х в молекулах MXk тем прочнее, чем больше перекрывание электронных облаков гибридных орбиталей атомов М и X, то есть чем сильнее эти облака сконцентрированы вдоль направлений М - X. Поэтому молекулы MXk должны иметь такое геометрическое строение, при котором плотность гибридных АО вдоль направлений связей максимальна, а валентные углы Х - М - Х совпадают с углами между направлениями гибридных АО центрального атома. Например, в молекулах типа PH3 и SH2 связи осуществляются почти чистыми 3р-орбиталями центральных атомов, и поэтому PH3 и SH2 имеют пирамидальное и угловое строение с углами Н - М - Н Валентность 90°. В дигалогенидах Zn, Cd, Hg, двуокисях, дисульфидах и др. соединениях углерода и его аналогов связи образуются за счёт sp-гибридных АО с валентным углом 180°, так что все молекулы типа CdCl2, Hg (CH3)2, HgI2, CS2, SiO2 и др. в парах имеют линейное строение. В случае Са, Sr, Вa, Ra и переходных металлов III-VI групп смешанная гибридизация sp + sd приводит к тому, что молекулы типа CaF2, SrF2, BaHal2, TiO2, HfO2, TaO2, ThO2, UO2 и др. имеют угловое строение.
С проблемой В. тесно связано приближённое понятие валентного состояния атома- гипотетического состояния, в котором находится атом в молекуле. Оно характеризуется валентной конфигурацией, то есть типом и числом заполненных и пустых валентных АО; их гибридизацией, воспроизводящей геометрическое строение ближайшего окружения рассматриваемого атома; числом электронов (в теории локализованных пар - это целое число: 2, 1 или 0), заселяющих каждую из гибридных АО, и относительной ориентацией спинов электронов. Например, в молекуле метана CH
4 атом С (см.
рис. 4) имеет валентную конфигурацию 2
s2
p3 с четырьмя тетрагональными
sp3-гибридными орбиталями (
te), направленными к вершинам тетраэдра, каждая из которых заселена одним электроном с неопределенно ориентированным спином, осуществляющим одну гайтлер-лондоновскую связь с соответствующим атомом Н. Как правило, валентное состояние атома в молекуле не совпадает с основным состоянием изолированного атома. Так, у углерода и его аналогов основное состояние (
рис. 4, а) может быть лишь двухвалентным. У всех атомов II группы периодической системы основное состояние
s2 вообще не может быть валентным, и для образования молекул типа ZnCl и ZnCl
2 необходимо возбуждение
s-электрона на ближайший пустой
р-уровень. Энергия возбуждения валентного состояния из основного состояния для разных атомов различна и может достигать нескольких сотен
ккал/моль, давая существенный вклад в общий энергетический баланс образования молекул из атомов. В случае Zn, Cd и Hg возбуждение
s →
р происходит при присоединении первого атома галогена и требует значительных затрат энергии (90-120
ккал/моль), поэтому энергия разрыва связи М - Hal в двухатомных молекулах MHal значительно меньше, чем связи HalM - Hal в трёхатомных молекулах MHal
2 (см.
Энергия химической связи)
. У Ca, Sr, Вa, Ra затраты на возбуждение s →
р или
s →
d значительно меньше (30-50
ккал/моль), и здесь энергии разрыва связей в молекулах галогенидов гораздо ближе друг другу.
В комплексных соединениях координационное число центрального атома часто больше числа электронов в его валентной оболочке. Важную роль здесь играют
Донорно-акцепторная связь и дативные связи, образующиеся за счёт неподелённой электронной пары (то есть пары электронов с противоположными спинами, занимающих одну АО) одного атома и пустой орбитали другого. Соответственно должны быть расширены и представления о В.: способность к образованию связей, а следовательно и В. атома, обусловливается не только неспаренными электронами, но и неподелёнными парами и пустыми орбиталями валентной оболочки. Наибольшая суммарная В. должна быть равна числу всех АО, составляющих валентную оболочку атома, поскольку каждая валентная АО, независимо от того, сколькими электронами она заселена у атома в валентном состоянии, потенциально способна образовать одну связь (гайтлер-лондоновскую, донорно-акцепторную или дативную). В рамках этой концепции максимальная В. всех элементов второго периода от Li до F равна 4 (одна
s-opбиталь + три
р-орбитали), у элементов следующих периодов - 9 (за счёт ещё пяти
d-opбиталей) и т.д. Решение же вопроса о том, какие из этих четырёх или девяти В. насыщаются и какие остаются неиспользованными, в соединениях каждого конкретного типа определяется не только свойствами самого атома и его положением в периодической системе, но и особенностями соединения в целом. Полный ответ на него может быть получен с помощью квантово-химических расчётов. За счёт донорно-акцепторного взаимодействия фактическое число связей атома (а следовательно и его В.) в комплексных и даже в простых соединениях в общем случае может быть больше не только числа его неспаренных электронов, но и числа связанных с ним соседних атомов.
Следует помнить, что подразделение связей в соединениях на гайтлер-лондоновские, донорно-акцепторные и дативные имеет, вообще говоря, лишь генетический смысл, поскольку после того как соединение образуется, в нём происходит перераспределение электронной плотности и выравнивание связей: например, в каждом из комплексных анионов типа [BF4]-, [BeF4]2-, [SiFe6] 2-, [АlF6]3-, [ZnF6]4- и др. все связи М - F совершенно одинаковы.
Установлено также, что в солях ион NO
3- имеет структуру правильного треугольника, а ионы
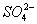
и
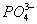
- структуру правильного тетраэдра. Поэтому строение молекул соответствующих солей точнее описывается приведёнными на
рис. 5 структурными формулами
г - е, а не традиционными формулами
а - в,
которые не учитывают реальной структуры ионов.
Теория локализованных пар ограничена в основном несопряжёнными органическими и простыми неорганическими соединениями. Так, в случае "электронно-избыточных" молекул типа PF5, SF6, IF7, XeF6 эта теория не может объяснить осуществления высших В. у атомов Р, S, I, Xe без привлечения валентных состояний с большими целочисленными заселённостями внешних d-opбиталей (sp3d для Р, sp3d3 для I, s2p3d3 для Xe и т.д.); однако энергии возбуждения последних столь велики (200-400 ккал/моль и более), что затраты на их возбуждение вряд ли могут окупиться за счёт выигрыша в энергии при образовании связей. Аналогичные трудности возникают при рассмотрении комплексных соединений, координационных кристаллов и т.д. В "электронно-дефицитных" молекулах типа В2Н6 (рис. 1, в) число связей, образуемых атомом Н, больше числа имеющихся у него валентных АО, так что связи мостиковых Н с двумя атомами В могут быть описаны только трёхцентровыми молекулярными орбиталями, охватывающими фрагменты В - Н - В. В случае ароматических и сопряжённых молекул типа C5H5, C6H6, C7H7 и др., их комплексов с металлами (рис. 3) и других производных валентные 2рπ-электроны в равной степени принадлежат всем атомам С и могут быть описаны лишь с помощью делокализованных молекулярных орбиталей, охватывающих всё кольцо или углеродный остов в целом. Иными словами, представления о локализованных В. и связях оказались слишком узкими, чтобы вместить все известные типы соединений.
Поэтому естественным следующим шагом в развитии общей теории В. стал метод молекулярных орбиталей, MO, который рассматривает молекулу как совокупность ядер и электронов, где каждый электрон движется в поле остальных электронов и всех ядер. Молекулярные орбитали, описывающие состояние электронов, в общем случае охватывают все атомы молекулы, так что каждый атом способен в принципе образовывать связи со всеми остальными атомами молекулы. Метод МО значительно более общ и последователен, что делает его в принципе пригодным для описания любых классов соединений. (См.
Молекулярных орбиталей метод,
Химическая связь.)
Лит.: Сыркин Я. К., Периодическая система и проблема валентности, М., 1971; Сыркин Я. К. и Дяткина М. Е., Химическая связь и строение молекул, М.-Л., 1946; Паулинг Л., Природа химической связи, пер. с англ., М. - Л., 1947; Шусторович Е. М., Новое в учении о валентности, М., 1968; Коулсон Ч., Валентность, пер. с англ., М., 1965: Маррел Д., Кеттл С., Теддер Д., Теория валентности, пер. с англ., М., 1968; Астахов К. В., Современное состояние периодической системы Д. И. Менделеева, М., 1969.
О. П. Чаркин.
Под редакцией академика Я. К. Сыркина.
Рис. 1. Мостиковые лиганды (Cl, H, CH3) в димерных и полимерных соединениях: Al2Cl6 (а), (PdCl2)x (б), В2Н6 (в) и Al2(CH3)6 (г).
Рис. 2. Кластерная структура Mo
6Cl
4+8 (

- атом Mo,
- атом Cl).
Рис. 3. (π-Комплексы переходных металлов (
- атом металла,
- атом С): ферроцен Fe(C
5H
5)
2 (а), дибензолхром Cr(C
6H
6)
2 (б).
Рис. 4. Схема возбуждения валентного состояния (г) атома углерода в молекуле типа CH4 из основного состояния (а): а - основное состояние наинизшей конфигурации 2s22p2; б - нижнее состояние валентной конфигурации 2s2p3; в- гибридизация АО; г-неопределённая ориентация спинов валентных электронов (валентное состояние).
Рис. 5. Структурные формулы молекул: NaNO3 (а, г), Na2SO4 (б, д) и Na3PO4 (в, е).
II
Вале́нтность
в языкознании, потенциальная сочетаемость языковых элементов (фонемы, морфемы, слова и т.д.), определяющая способность вступать в комбинации с другими языковыми элементами, преимущественно того же уровня. В. является категориальной или индивидуальной в зависимости от того, подразумевается ли сочетаемость целых классов слов (например, глаголов-сказуемых с существительными-подлежащими) или отдельных слов (например слова "знобит" с одушевлёнными существительными в винительном падеже). Если при функционировании элемента В. непременно реализуется, то она называется обязательной. При простой В. элемент сочетается с одним элементом, при комплексной (многоместной) В. - одновременно с несколькими (например, сказуемое употребляется одновременно с подлежащим, дополнением и обстоятельством). Тождество В. является одним из оснований для объединения элементов в один класс. Исчисление В. языковых элементов, особенно на синтаксическом уровне, - распространённый метод формального анализа языка.
Лит.: Лейкина Б. М., Некоторые аспекты характеристики валентностей, в сборнике: Доклады на конференции по обработке информации, машинному переводу и автоматическому чтению текста, в. 5, М., 1961.
В. В. Раскин.


![Структурная формула молекулы [[этан]]а](https://commons.wikimedia.org/wiki/Special:FilePath/Oxidationszahlen Ethan.svg?width=200 "Структурная формула молекулы [[этан]]а")



