Введите слово или словосочетание на любом языке 👆
Язык:
Перевод и анализ слов искусственным интеллектом ChatGPT
На этой странице Вы можете получить подробный анализ слова или словосочетания, произведенный с помощью лучшей на сегодняшний день технологии искусственного интеллекта:
- как употребляется слово
- частота употребления
- используется оно чаще в устной или письменной речи
- варианты перевода слова
- примеры употребления (несколько фраз с переводом)
- этимология
Что (кто) такое Спектральный анализ рентгеновский - определение
Дисперсионный рентгеновский микроанализ; Рентгеновский спектральный анализ; Рентгеновская спектроскопия; Спектральный анализ рентгеновский; Энергодисперсионный рентгеноспектральный микроанализ; Спектроскопия, рентгеновская
![кристаллографической плоскостью]] <math>10\bar{1}1.</math> Римскими цифрами I, II, III отмечены дифракционные спектры 1-го, 2-го и 3-го порядков.](https://commons.wikimedia.org/wiki/Special:FilePath/Molybdenum specimen chart-int.svg?width=200 "кристаллографической плоскостью]] <math>10\bar{1}1.</math> Римскими цифрами I, II, III отмечены дифракционные спектры 1-го, 2-го и 3-го порядков.")
Найдено результатов: 371
Спектральный анализ рентгеновский
элементный анализ вещественного состава материалов по их рентгеновским спектрам (См. Рентгеновские спектры). Качеств. С. а. р. выполняют по спектральному положению характеристических линий в спектре испускания исследуемого образца, его основой является Мозли закон; количественный С. а. р. осуществляют по интенсивностям этих линий. Методами С. а. р. могут быть определены все элементы с атомным номером Z ≥ 12 (в некоторых случаях - и более лёгкие). Порог чувствительности С. а. р. в большинстве случаев Спектральный анализ рентгеновский 10-2-10-4 \%, продолжительность его (вместе с подготовкой пробы) несколько мин. С. а. р. не разрушает пробу.
Наиболее распространённый вид С. а. р. - анализ валового состава материалов по их флуоресцентному рентгеновскому излучению. Выполняется он по относительной интенсивности линий, которая измеряется с высокой точностью спектральной аппаратурой рентгеновской (См. Спектральная аппаратура рентгеновская). Относительная точность количественного С. а. р. колеблется от 0,3 до 10\% в зависимости от состава пробы; на интенсивность аналитической линии каждого элемента влияют все остальные элементы пробы. Поэтому одной и той же измеренной интенсивности I1 аналитической линии i могут соответствовать различные концентрации C1, C2, С3, ... определяемого элемента (см. рис.) в зависимости от наполнителя - состава пробы за исключением определяемого элемента. Вследствие этого т. н. вырождения интенсивности по концентрации С. а. р. возможен лишь на основе общей теории зависимости li от концентраций всех n компонентов пробы - системы n уравнений связи.
На основе общей теории анализа разработано несколько частных методов. При отсутствии в пробе мешающих элементов можно применять простейший из них - метод внешнего стандарта: измерив интенсивность аналитической линии пробы, по аналитическому графику образца известного состава (стандарта) находят концентрацию исследуемого элемента. Для многокомпонентных проб иногда применяют метод внутреннего стандарта, в котором ординатой аналитического графика служит отношение интенсивностей линий определяемого элемента и внутреннего стандарта - добавленного в пробу в известном количестве элемента, соседнего (в периодической системе элементов) с определяемым. Во многих случаях успешно применяют метод добавок в пробу в известном количестве определяемого элемента или наполнителя. По изменению интенсивности аналитической линии можно найти первоначальную концентрацию определяемого элемента.
В промышленности применяют метод стандарта-фона, в котором ординатой аналитического графика является отношение интенсивности аналитической линии флуоресцентного излучения образца и близкой к ней линии первичного рентгеновского излучения, рассеянного пробой. Это отношение во многих случаях мало зависит от состава наполнителя. Для анализа сложных многокомпонентных проб полную систему уравнений связи расшифровывают на ЭВМ по методу последовательных (обычно трёх-четырёх) приближений.
С. а. р. валового состава нашёл применение на обогатительных фабриках цветной металлургии - для контрольных целей и для экспрессного анализа; на металлургических заводах - для определения потерь металла в шлаках, маркировки сплавов сложного состава, контроля состава латуней в процессе плавки и т. д.; на цементных заводах - для контроля состава цементно-сырьевых смесей. Валовый С. а. р. применяется также для силикатного анализа.
Рентгеновский микроанализ (локальный анализ) участков пробы Спектральный анализ рентгеновский 1-3 мкм2 (т. е. меньше размеров зерна сплава) выполняют с помощью электронно-зондового микроанализатора по рентгеновскому спектру исследуемого участка. Он требует точного введения поправок на атомный номер определяемого элемента, поглощение его излучения в пробе и его флуоресценцию, возбуждаемую тормозной компонентой излучения и характеристическим излучением др. элементов пробы.
Микроанализ применяют при исследовании взаимной диффузии двух- и трёх-компонентных систем; процессов кристаллизации (См. Кристаллизация) (по дендритной ликвации, сегрегации примесных атомов на дислокациях основного компонента, концентрации некоторых фаз на границе зёрен); локальных флуктуаций состава плохо гомогенизированных сплавов и пр.
Лит.: Блохин М. А., Методы рентгено-спектральных исследований, М., 1959; Блохин М. А., Ильин Н. П., Рентгеноспектральный анализ, "Журнал аналитической химии", 1967, т. 22, в. 11; Лосев Н. Ф., Количественный рентгеноспектральный флуоресцентный анализ, М., 1969; Плотников Р. И., Пшеничный Г. А.,
флюоресцентный рентгенорадиометрический анализ, М., 1973; Бирке Л. С., Рентгеновский микроанализ с помощью электронного зонда, пер. с англ., М., 1966; Физические основы рентгеноспектрального локального анализа, пер. с англ., М., 1973; Электронно-зондовый микроанализ, пер. с англ., М., 1974.
М. А. Блохин.
Графики зависимости интенсивности li аналитич. линии i от концентрации С определяемого элемента (аналитические графики) для случаев, когда поглощение наполнителя меньше (1), равно (2) или больше (3) поглощения определяемого элемента, Iф - интенсивность фона.
Рентгеноспектральный анализ
Рентгеноспектра́льный ана́лиз — инструментальный метод элементного анализа, основанный на изучении спектра рентгеновских лучей, прошедших сквозь образец или испущенных им.
Рентгеновская спектроскопия
получение рентгеновских спектров (См. Рентгеновские спектры) испускания и поглощения и их применение к исследованию электронной энергетической структуры атомов, молекул и твёрдых тел. К Р. с. относят также рентгено-электронную спектроскопию, т. е. спектроскопию рентгеновских фото- и оже-электронов, исследование зависимости интенсивности тормозного и характеристического спектров от напряжения на рентгеновской трубке (См. Рентгеновская трубка) (метод изохромат), спектроскопию потенциалов возбуждения.
Рентгеновские спектры испускания получают либо бомбардировкой исследуемого вещества, служащего мишенью в рентгеновской трубке, ускоренными электронами (первичные спектры), либо облучением вещества первичными лучами (флуоресцентные спектры). Спектры испускания регистрируются рентгеновскими спектрометрами (см. Спектральная аппаратура рентгеновская). Их исследуют по зависимости интенсивности излучения от энергии рентгеновского фотона. Форма и положение рентгеновских спектров испускания дают сведения об энергетическом распределении плотности состояний валентных электронов, позволяют экспериментально выявить симметрию их волновых функций и их распределение между сильно связанными локализованными электронами атома и коллективизированными электронами твёрдого тела.
Рентгеновские спектры поглощения образуются при пропускании узкого участка спектра тормозного излучения через тонкий слой исследуемого вещества. Исследуя зависимость коэффициента поглощения рентгеновского излучения веществом от энергии рентгеновских фотонов, получают сведения об энергетическом распределении плотности свободных электронных состояний. Спектральные положения границы спектра поглощения и максимумов его тонкой структуры позволяют найти кратность зарядов ионов в соединениях (её можно определить во многих случаях и по смещениям основных линий спектра испускания). Р. с. даёт возможность также установить симметрию ближнего окружения атома, исследовать природу химической связи. Рентгеновские спектры, возникающие при бомбардировке атомов мишени тяжёлыми ионами высокой энергии, дают информацию о распределении излучающих атомов по кратности внутренних ионизаций. Рентгеноэлектронная спектроскопия находит применение для определения энергии внутренних уровней атомов, для химического анализа и определения валентных состояний атомов в химических соединениях.
Лит.: Блохин М. А., Физика рентгеновских лучей, М., 1957; Рентгеновские лучи, под ред. М. А. Блохина, М., 1960; Баринский Р. Л., Нефедов В. И., Рентгено-спектральное определение заряда атомов в молекулах, М., 1966; Зимкина Т. М., Фомичев В. А., Ультрамягкая рентгеновская спектроскопия, Л, 1971; Немошкаленко В. В., Рентгеновская эмиссионная спектроскопия металлов и сплавов, К., 1972; X-ray spectroscopy, ed. L. V. Azaroff, N. - Y., 1974.
М. А. Блохин.
РЕНТГЕНОВСКАЯ СПЕКТРОСКОПИЯ
методы исследования атомной структуры по рентгеновским спектрам. Для получения рентгеновских спектров исследуемое вещество бомбардируют электронами в рентгеновской трубке либо возбуждают флуоресценцию исследуемого вещества, облучая его рентгеновским излучением.
Рентгенофлуоресцентный анализ

Рентгенофлуоресцентный анализ (РФА) — один из современных спектроскопических методов исследования вещества с целью получения его элементного состава, то есть его элементного анализа. С помощью него могут быть найдены различные элементы от бериллия (Be) до урана (U).
Спектральный анализ

СТРАНИЦА ЗНАЧЕНИЙ В ПРОЕКТЕ ВИКИМЕДИА
Спектральный анализ (физич., химич.); Спектроскопический анализ
I
Спектра́льный ана́лиз
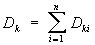

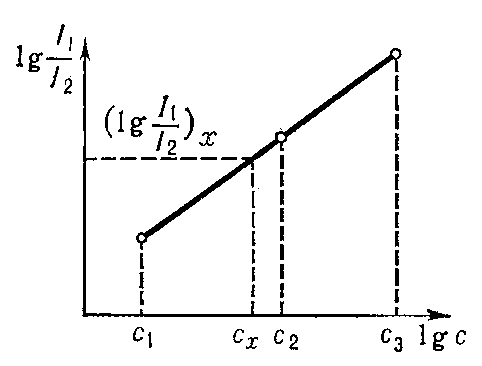


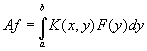 ,
,
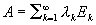
 (*)
(*)


физический метод качественного и количественного определения атомного и молекулярного состава вещества, основанный на исследовании его спектров. Физическая основа С. а.- Спектроскопия атомов и молекул, его классифицируют по целям анализа и типам спектров (см. Спектры оптические). Атомный С. а. (АСА) определяет элементный состав образца по атомным (ионным) спектрам испускания и поглощения, молекулярный С. а. (МСА) - молекулярный состав веществ по молекулярным спектрам поглощения, люминесценции (См. Люминесценция) и комбинационного рассеяния света (См. Комбинационное рассеяние света).
Эмиссионный С. а. производят по спектрам испускания атомов, ионов и молекул, возбуждённым различными источниками электромагнитного излучения в диапазоне от γ-излучения до микроволнового. Абсорбционный С. а. осуществляют по спектрам поглощения электромагнитного излучения анализируемыми объектами (атомами, молекулами, ионами вещества, находящегося в различных агрегатных состояниях).
Историческая справка. В основе АСА лежит индивидуальность спектров испускания и поглощения химических элементов, установленная впервые Г. Р. Кирхгофом и Р. Бунзеном (1859-61). В 1861 Кирхгоф доказал на основе этого открытия присутствие в хромосфере Солнца ряда элементов, положив начало астрофизике. В 1861-1923 с помощью АСА было открыто 25 элементов. В 1932 спектральным методом был открыт дейтерий.
Высокая чувствительность и возможность определения многих элементов в пробах малой массы сделали АСА эффективным методом качественного анализа элементного состава объектов. В 1926 нем. физик В. Герлах положил начало количественному С. а. Для развития С. а. и внедрения его на промышленных предприятиях СССР большую роль сыграли Г. С. Ландсберг, С. Л. Мандельштам, А. К. Русанов (Москва), А. Н. Филиппов, В. К. Прокофьев (Ленинград) и др.
Атомный спектральный анализ (АСА)
Эмиссионный АСА состоит из следующих основных процессов:
1) отбор представительной пробы, отражающей средний состав анализируемого материала или местное распределение определяемых элементов в материале;
2) введение пробы в источник излучения, в котором происходят испарение твёрдых и жидких проб, диссоциация соединений и возбуждение атомов и ионов;
3) преобразование их свечения в спектр и его регистрация (либо визуальное наблюдение) с помощью спектрального прибора (См. Спектральные приборы);
4) расшифровка полученных спектров с помощью таблиц и атласов спектральных линий элементов.
На этой стадии заканчивается качественный АСА. Наиболее результативно использование чувствительных (т. н. "последних") линий, сохраняющихся в спектре при минимальной концентрации определяемого элемента. Спектрограммы просматривают на измерительных микроскопах, компараторах, спектропроекторах. Для качественного анализа достаточно установить наличие или отсутствие аналитических линий определяемых элементов. По яркости линий при визуальном просмотре можно дать грубую оценку содержания тех или иных элементов в пробе.
Количественный АСА осуществляют сравнением интенсивностей двух спектральных линий в спектре пробы, одна из которых принадлежит определяемому элементу, а другая (линия сравнения) - основному элементу пробы, концентрация которого известна, или специально вводимому в известной концентрации элементу ("внутреннему стандарту").
В основе количественного АСА лежит соотношение, связывающее концентрацию с определяемого элемента с отношением интенсивностей линии определяемой примеси (I1) и линии сравнения (I2):
I1/I2 = acb
(постоянные а и b определяются опытным путём), или
lg(I1/I2) = b lgс + lga.
С помощью стандартных образцов (не менее 3) можно построить график зависимости lg(I1/I2.) от lg с (градуировочный график, рис. 1) и определить по нему а и b. Значения I1 и I2 можно получать непосредственно путём фото-электрической регистрации или путём фотометрирования (измерения плотности почернения) линии определяемой примеси и линии сравнения при фоторегистрации. Фотометрирование производят на Микрофотометрах.
Для возбуждения спектра в АСА используют различные источники света и соответственно различные способы введения в них образцов. Выбор источника зависит от конкретных условий анализа определённых объектов. Тип источника и способ введения пробы составляют главное содержание частных методик АСА.
Первым искусственным источником света в АСА было пламя газовой горелки - источник весьма удобный для быстрого и точного определения многих элементов. Температура пламён горючих газов не высока (от 2100 К для смеси водород - воздух до 4500 К для редко используемой смеси кислород - циан). С помощью фотометрии пламени определяют около 70 элементов по их аналитическим линиям, а также по молекулярным полосам соединений, образующихся в пламёнах.
В эмиссионном АСА широко используют электрические источники света. В электрической дуге постоянного тока между специально очищенными угольными электродами различной формы, в каналы которых помещают исследуемое вещество в измельченном состоянии, можно производить одновременное определение десятков элементов. Она обеспечивает относительно высокую температуру нагрева электродов и благоприятные условия возбуждения атомов пробы в дуговой плазме, однако точность этого метода невысока из-за нестабильности разряда. Повышая напряжение до 300-400 в или переходя к высоковольтной дуге (3000-4000 в), можно увеличить точность анализа.
Более стабильные условия возбуждения создаёт дуга переменного тока. В современных генераторах дуги переменного тока (см., напр., рис. 2) можно получить различные режимы возбуждения: низковольтную искру, высокочастотную искру, дугу переменного тока, импульсный разряд и т. д. Такие источники света с различными режимами используют при определении металлов и трудновозбудимых элементов (углерод, галогены, газы, содержащиеся в металлах, и т. д.). Высоковольтная конденсированная искра (рис. 3) служит главным образом источником света при анализе металлов. Стабильность искрового разряда позволяет получать высокую воспроизводимость анализа, однако сложные процессы, происходящие на поверхностях анализируемых электродов, приводят к изменениям состава плазмы разряда. Чтобы устранить это явление, приходится производить предварительный обжиг проб и нормировать форму и размеры проб и стандартных образцов.
В АСА перспективно применение стабилизированных форм электрического разряда типа плазмотронов различных конструкций, высокочастотного индукционного разряда, СВЧ-разряда, создаваемого магнетронными генераторами, высокочастотного факельного разряда. С помощью различных приёмов введения анализируемых веществ в плазму этих типов разряда (продувка порошков, распыление растворов и т. д.) значительно повышена относительная точность анализа (до 0,5-3\% ), в том числе и компонентов сложных проб, содержание которых составляет десятки \%. В некоторых важных случаях анализа чистых веществ применение этих типов раз ряда снижает пределы определения примесей на 1-2 порядка (до 10-5-10-6 \% ).
Для анализа чистых веществ, радиоактивных материалов, смесей газов, изотопного анализа, спектрально-изотопного определения газов в металлах и твёрдых веществах и т. д. весьма перспективным оказалось использование разряда в полом катоде и безэлектродных ВЧ-и СВЧ-разрядов. В АСА в качестве источников возбуждения применяются также лазеры (см. Спектроскопия лазерная).
Атомно-абсорбционный С. а. (ААА) и атомно-флуоресцентный С. а. (АФА). В этих методах пробу превращают в пар в атомизаторе (пламени, графитовой трубке, плазме стабилизированного ВЧ-или СВЧ-разряда). В ААА свет от источника дискретного излучения, проходя через этот пар, ослабляется и по степени ослабления интенсивностей линий определяемого элемента судят о концентрации его в пробе. ААА проводят на специальных Спектрофотометрах. Методика проведения ААА по сравнению с др. методами значительно проще, для него характерна высокая точность определения не только малых, но и больших концентраций элементов в пробах. ААА с успехом заменяет трудоёмкие и длительные химические методы анализа, не уступая им в точности .
В АФА атомные пары пробы облучают светом источника резонансного излучения и регистрируют флуоресценцию определяемого элемента. Для некоторых элементов (Zn, Cd, Hg и др.) относительные пределы их обнаружения этим методом весьма малы (Спектральный анализ10-5-106 \%).
АСА позволяет проводить измерения изотопного состава. Некоторые элементы имеют спектральные линии с хорошо разрешенной структурой (например, Н, Не, U). Изотопный состав этих элементов можно измерять на обычных спектральных приборах с помощью источников света, дающих тонкие спектральные линии (полый катод, безэлектродные ВЧ-и СВЧ-лампы). Для проведения изотопного спектрального анализа большинства элементов требуются приборы высокой разрешающей способности (например, эталон Фабри - Перо). Изотопный спектральный анализ можно также проводить по электронно-колебательным спектрам молекул, измеряя изотопные сдвиги полос, достигающие в ряде случаев значительной величины.
Экспрессные методы АСА широко применяются в промышленности, сельском хозяйстве, геологии и многих др. областях народного хозяйства и науки. Значительную роль АСА играет в атомной технике, производстве чистых полупроводниковых материалов, сверхпроводников и т. д. Методами АСА выполняется более 3/4 всех анализов в металлургии. С помощью квантометров проводят оперативный (в течение 2-3 мин) контроль в ходе плавки в мартеновском и конвертерном производствах. В геологии и геологической разведке для оценки месторождений производят около 8 млн. анализов в год. АСА применяется для охраны окружающей среды и анализа почв, в криминалистике и медицине, геологии морского дна и исследовании состава верхних слоев атмосферы, при разделении изотопов и определении возраста и состава геологических и археологических объектов и т. д.
Лит.: Заидель А. Н., Основы спектрального анализа, М., 1965; Методы спектрального анализа, М,, 1962; Эмиссионный спектральный анализ атомных материалов, Л. - М., 1960; Русанов А. К., Основы количественного спектрального анализа руд и минералов. М., 1971; Спектральный анализ чистых веществ, под ред. X. И. Зильберштейна, [Л.], 1971; Львов Б. В., Атомно-абсорбционный спектральный анализ, М., 1966; Петров А. А., Спектрально-изотопный метод исследования материалов, Л., 1974; Тарасевич Н. И.. Семененко К. А., Хлыстова А. Д., Методы спектрального и химико-спектрального анализа, М., 1973: Прокофьев В. К., Фотографические методы количественного спектрального анализа металлов и сплавов, ч. 1-2, М. - Л., 1951; Менке Г., Менке Л., Введение в лазерный эмиссионный микроспектральный анализ, пер. с нем., М., 1968; Королев Н. В., Рюхин В. В., Горбунов С. А., Эмиссионный спектральный микроанализ, Л., 1971; Таблицы спектральных линий, 3 изд., М., 1969; Стриганов A. P., Свентицкий Н. С., Таблицы спектральных линий нейтральных и ионизованных атомов, М., 1966.
Л. В. Липис.
Молекулярный спектральный анализ (МСА)
В основе МСЛ лежит качественное и количественное сравнение измеренного спектра исследуемого образца со спектрами индивидуальных веществ. Соответственно различают качественный и количественный МСА. В МСА используют различные виды молекулярных спектров (См. Молекулярные спектры), вращательные [спектры в микроволновой и длинноволновой инфракрасной (ИК) областях], колебательные и колебательно-вращательные [спектры поглощения и испускания в средней ИК-области, спектры комбинационного рассеяния света (КРС), спектры ИК-флуоресценции], электронные, электронно-колебательные и электронно-колебательно-вращательные [спектры поглощения и пропускания в видимой и ультрафиолетовой (УФ) областях, спектры флуоресценции]. МСА позволяет проводить анализ малых количеств (в некоторых случаях доли мкг и менее) веществ, находящихся в различных агрегатных состояниях.
Основные факторы, определяющие возможности методов МСА:
1) информативность метода. Условно выражается числом спектрально разрешаемых линий или полос в определённом интервале длин волн или частот исследуемого диапазона (для микроволнового диапазона оно Спектральный анализ 105, для средней ИК-области в спектрах твёрдых и жидких веществ Спектральный анализ 103);
2) количество измеренных спектров индивидуальных соединений;
3) существование общих закономерностей между спектром вещества и его молекулярным строением;
4) чувствительность и избирательность метода;
5) универсальность метода;
6) простота и доступность измерений спектров.
Качественный МСА устанавливает молекулярный состав исследуемого образца. Спектр молекулы является его однозначной характеристикой. Наиболее специфичны спектры веществ в газообразном состоянии с разрешенной вращательной структурой, которые исследуют с помощью спектральных приборов высокой разрешающей способности. Наиболее широко используют спектры ИК-поглощения и КРС веществ в жидком и твёрдом состояниях, а также спектры поглощения в видимой и УФ-областях. Широкому внедрению метода КРС способствовало применение для их возбуждения лазерного излучения.
Для повышения эффективности МСА в некоторых случаях измерение спектров комбинируют с др. методами идентификации веществ. Так, всё большее распространение получает сочетание хроматографического разделения смесей веществ с измерением ИК-спектров поглощения выделенных компонент.
К качественному МСА относится также т. н. структурный молекулярный анализ. Установлено, что молекулы, имеющие одинаковые структурные элементы, обнаруживают в спектрах поглощения и испускания общие черты. Наиболее ярко это проявляется в колебательных спектрах. Так, наличие сульфгидрильной группы (-SH) в структуре молекулы влечёт за собой появление в спектре полосы в интервале 2565-2575 см-1, нитрильная группа (-CN) характеризуется полосой 2200-2300 cм-1 и т. д. Присутствие таких характеристических полоса колебательных спектрах веществ с общими структурными элементами объясняется характеристичностью частоты и формы многих молекулярных колебаний. Подобные особенности колебательных (и в меньшей степени электронных) спектров во многих случаях позволяют определять структурный тип вещества.
Качественный анализ существенно упрощает и ускоряет применение ЭВМ. В принципе его можно полностью автоматизировать, вводя показания спектральных приборов непосредственно в ЭВМ. В её памяти должны быть заложены спектральные характеристические признаки многих веществ, на основании которых машина произведёт анализ исследуемого вещества.
Количественный МСА по спектрам поглощения основан на Бугера - Ламберта - Бера законе, устанавливающем связь между интенсивностями падающего и прошедшего через вещество I света от толщины поглощающего слоя I и концентрации вещества с:
I(l)=I0e-χcl
Коэффициент χ является характеристикой поглощающей способности определяемого компонента для данной частоты излучения. Важное условие проведения количественного МСА - независимость χ от концентрации вещества и постоянство χ в измеряемом интервале частот, определяемом шириной щели спектрофотометра. МСА по спектрам поглощения проводят преимущественно для жидкостей и растворов, для газов он значительно усложняется.
В практическом МСА обычно измеряют т. н. оптическую плотность:
D = In (/о//) = χсl.
Если смесь состоит из n веществ, не реагирующих друг с другом, то оптическая плотность смеси на частоте ν аддитивна: 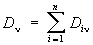 . Это позволяет проводить полный или частичный анализ многокомпонентных смесей. Задача в этом случае сводится к измерению значений оптической плотности в m точках спектра смеси (m ≥ n) и решению получаемой системы уравнений:
. Это позволяет проводить полный или частичный анализ многокомпонентных смесей. Задача в этом случае сводится к измерению значений оптической плотности в m точках спектра смеси (m ≥ n) и решению получаемой системы уравнений:
Для количественного МСА обычно пользуются спектрофотометрами, позволяющими производить измерение /(ν) в сравнительно широком интервале ν . Если полоса поглощения исследуемого вещества достаточно изолирована и свободна от наложения полос др. компонент смеси, исследуемый спектральный участок можно выделить, например, при помощи интерференционного Светофильтра. На его основе конструируют специализированные анализаторы, широко используемые в промышленности.
При количественном МСА по спектрам КРС чаще всего интенсивность линии определяемого компонента смеси сравнивают с интенсивностью некоторой линии стандартного вещества, измеренной в тех же условиях (метод "внешнего стандарта"). В др. случаях стандартное вещество добавляют к исследуемому в определённом количестве (метод "внутреннего стандарта" ).
Среди др. методов качественного и количественного МСА наибольшей чувствительностью обладает флуоресцентный анализ, однако в обычных условиях он уступает методам колебательной спектроскопии в универсальности и избирательности. Количественный МСА по спектрам флуоресценции основан на сравнении свечения раствора исследуемого образца со свечением ряда эталонных растворов близкой концентрации.
Особое значение имеет МСА с применением техники замороженных растворов в специальных растворителях, например парафинах (см. Шпольского эффект). Спектры веществ в таких растворах (спектры Шпольского) обладают ярко выраженной индивидуальностью, они резко различны для близких по строению и даже изомерных молекул. Это позволяет идентифицировать вещества, которые по спектрам их флуоресценции в обычных условиях установить не удаётся. Например, метод Шпольского даёт возможность осуществлять качественный и количественный анализ сложных смесей, содержащих ароматические углеводороды. Качественный анализ в этом случае производят по спектрам люминесценции и поглощения, количественный - по спектрам люминесценции методами "внутреннего" и "внешнего" стандартов. Благодаря исключительно малой ширине спектральных линий в спектрах Шпольского в этом методе удаётся достигнуть пороговой чувствительности обнаружения некоторых многоатомных ароматических соединений (Спектральный анализ 10Спектральный анализ11 г/см3).
Лит.: Чулановский В. М., Введение в молекулярный спектральный анализ, М. - Л., 1951; Беллами Л., Инфракрасные спектры сложных молекул, пер. с англ., М., 1963; Применение спектроскопии в химии, пер. с англ., М., 1959; Определение индивидуального углеводородного состава бензинов прямой гонки комбинированным методом, М., 1959; Юденфренд С., Флуоресцентный анализ в биологии и медицине, пер. с англ., М., 1965.
В. Т. Алексанян.
Рис. 1. Градуировочный график (метод трёх эталонов).
Рис. 2. Принципиальная схема дуги переменного тока двойного питания: А - амперметр; R1 и R2 - реостаты; Тр - повышающий трансформатор: К - катушка индуктивности; АП - аналитический промежуток; П - вспомогательный промежуток; C1 и С2 - конденсаторы.
Рис. 3. Схема генератора конденсированной искры с управляющим промежутком: АП - регулируемый аналитический промежуток, образованный ванадиевыми электродами; R1 - реостат; Тр - питающий трансформатор; С - конденсатор; L - катушка индуктивности; П - управляющий промежуток; R2 - блокирующее сопротивление.
II
Спектра́льный ана́лиз
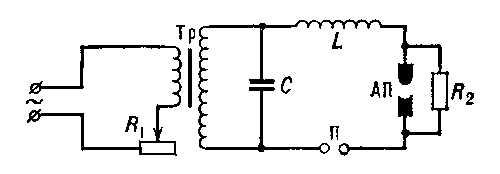
линейных операторов, обобщение выросшей из задач механики теории собственных значений (См. Собственные значения) и собственных векторов (См. Собственные векторы) матриц (т. е. линейных преобразований в конечномерном пространстве) на бесконечномерный случай (см. Линейный оператор, Операторов теория). В теории колебаний изучается движение системы с n степенями свободы в окрестности положения устойчивого равновесия, которое описывается системой линейных дифференциальных уравнений вида 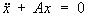 , где х есть n-мерный вектор отклонений обобщённых координат системы от их равновесных значений, а А - симметрическая положительно определённая матрица. Такое движение может быть представлено в виде наложения n гармонических колебаний (т. н. нормальных колебаний) с круговыми частотами, равными корням квадратным из всевозможных собственных значений λ k матрицы А. Нахождение нормальных колебаний системы здесь сводится к нахождению всех собственных значений λk; и собственных векторов xk матрицы А. Совокупность всех собственных значений матрицы называют её спектром. Если матрица А - симметрическая, то её спектр состоит из n действительных чисел λ1, ..., λn (некоторые из них могут совпадать друг с другом), а сама матрица с помощью перехода к новой системе координат может быть приведена к диагональному виду, т. е. отвечающее ей линейное преобразование А в n-мерном пространстве (т. н. самосопряжённое преобразование) допускает специальное представление - т. н. Спектральное разложение вида
, где х есть n-мерный вектор отклонений обобщённых координат системы от их равновесных значений, а А - симметрическая положительно определённая матрица. Такое движение может быть представлено в виде наложения n гармонических колебаний (т. н. нормальных колебаний) с круговыми частотами, равными корням квадратным из всевозможных собственных значений λ k матрицы А. Нахождение нормальных колебаний системы здесь сводится к нахождению всех собственных значений λk; и собственных векторов xk матрицы А. Совокупность всех собственных значений матрицы называют её спектром. Если матрица А - симметрическая, то её спектр состоит из n действительных чисел λ1, ..., λn (некоторые из них могут совпадать друг с другом), а сама матрица с помощью перехода к новой системе координат может быть приведена к диагональному виду, т. е. отвечающее ей линейное преобразование А в n-мерном пространстве (т. н. самосопряжённое преобразование) допускает специальное представление - т. н. Спектральное разложение вида
где E1,..., En - операторы проектирования на взаимно перпендикулярные направления собственных векторов х1, ......, xn. Несимметрическая же матрица А (которой отвечает несамосопряжённое линейное преобразование) имеет, вообще говоря, спектр, состоящий из комплексных чисел λ1, ..., λ1, и может быть преобразована лишь к более сложной, чем диагональная, жордановой форме [см. Нормальная (жорданова) форма матриц (См. Нормальная форма матриц)], отвечающей представлению линейного преобразования А, более сложному, чем описанное выше обычное спектральное разложение.
При изучении колебаний около состояния равновесия систем с бесконечным числом степеней свободы (например, однородной или неоднородной струны) задачу о нахождении собственных значений и собственных векторов линейного преобразования в конечномерном пространстве приходится распространить на некоторый класс линейных преобразований (т. е. линейных операторов) в бесконечномерном линейном пространстве. Во многих случаях (включая, в частности, и случай колебания струны) соответствующий оператор может быть записан в виде действующего в пространстве функций f(x) интегрального оператора А, так что здесь
где К(х, у) - заданная на квадрате а ≤ х, у ≤ b непрерывная функция двух переменных, удовлетворяющая условию симметрии К(х, у) = К(у, х). В этих случаях оператор А всегда имеет полную систему попарно ортогональных собственных функций (См. Собственные функции) φk, которым отвечает счётная последовательность действительных собственных значений λk, составляющих в своей совокупности спектр оператора А. Если рассматривать функции, на которые действует оператор А, как векторы гильбертова пространства, то действие А будет, как и в случае конечномерного самосопряжённого преобразования, сводиться к растяжению пространства вдоль системы взаимно ортогональных осей φk с коэффициентами растяжения λk (при λk < 0 такое растяжение имеет смысл растяжения с коэффициентом |λk|, объединённого с зеркальным отражением), а сам оператор А здесь снова будет иметь спектральное разложение вида
где Ek - операторы проектирования на направления φk.
С. а., развитый первоначально для интегральных операторов с симметричным ядром К(х, у), определённым и непрерывным в некоторой ограниченной области, был затем в рамках общей теории операторов распространён на многие другие типы линейных операторов (например, на интегральные операторы с ядром, имеющим особенность или заданным в неограниченной области, дифференциальные операторы в пространствах функций одного или нескольких переменных и т. д.), а также на абстрактно заданные линейные операторы в бесконечномерных линейных пространствах. Оказалось, однако, что такое распространение связано с существенным усложнением С. а., так как для многих линейных операторов собственные значения и собственные функции, понимаемые в обычном смысле, вообще не существуют. Поэтому в общем случае спектр приходится определять не как совокупность собственных значений оператора А, а как совокупность тех значений, для которых оператор (А - λЕ)-1, где Е - тождественный (единичный) оператор, не существует, или определён лишь на неплотном множестве, или является неограниченным оператором. Все собственные значения оператора принадлежат его спектру и в совокупности образуют его дискретный спектр; остальную часть спектра часто называют непрерывным спектром оператора [иногда же непрерывным спектром называют лишь совокупность тех λ, при которых оператор (А - λЕ)-1 определён на плотном множестве элементов пространства, но неограничен, а все точки спектра, не входящие ни в дискретный, ни в непрерывный спектр, называют остаточным спектром].
Наиболее разработан С. а. самосопряжённых линейных операторов в гильбертовом пространстве (обобщающих симметрические матрицы) и унитарных линейных операторов в том же пространстве (обобщающих унитарные матрицы). Самосопряжённый оператор А в гильбертовом пространстве всегда имеет чисто действительный спектр (дискретный, непрерывный или смешанный) и допускает спектральное разложение вида
где E(λ) - т. н. разложение единицы (отвечающее оператору А), т. е. семейство проекционных операторов (См. Проекционный оператор), удовлетворяющее специальным условиям. Точками спектра в данном случае являются точки роста операторной функции Е(λ); в случае чисто дискретного спектра все они являются скачками Е(λ), так что здесь
и спектральное разложение (*) сводится к разложению
Унитарный оператор в гильбертовом пространстве имеет спектр, расположенный на окружности |λ| = 1, и допускает спектральное разложение родственного (*) вида, но с заменой интегрирования от -∞ до ∞ интегрированием по этой окружности. Изучен также специальный класс нормальных операторов в гильбертовом пространстве, представимых в аналогичном представлению (*) виде, но где уже интегрирование в правой части распространено на более общее множество точек λ комплексной плоскости, представляющее собой спектр А. Что касается С. а. несамосопряжённых и не являющихся нормальными линейных операторов, обобщающих произвольные несимметрические матрицы, то ему были посвящены многочисленные работы Дж. Биркгофа (США), Т. Карлемана (Швеция), М. В. Келдыша, М. Г. Крейна (СССР), Б. Сёкефальви-Надя (Венгрия), Н. Данфорда (США) и многих др. учёных, но тем не менее соответствующая теория ещё далека от полной завершённости.
С. а. линейных операторов имеет целый ряд важных применений в классической механике (особенно теории колебаний), электродинамике, квантовой механике, теории случайных процессов, дифференциальных и интегральных уравнений и др. областях математики и математической физики.
Лит.: Курант P., Гильберт Д., Методы математической физики, пер. с нем., 3 изд., т. 1, М. - Л., 1951; Ахиезер Н. И., Глазман И.М., Теория линейных операторов в гильбертовом пространстве, 2 изд., М., 1966; Плеснер А. И., Спектральная теория линейных операторов, М., 1965; Рисе Ф., Секефальви Надь Б., Лекции по функциональному анализу, пер. с франц., М., 1954; Секефальви-Надь Б., Фояш Ч., Гармонический анализ операторов в гильбертовом пространстве, пер. с франц., М., 1970; Данфорд Н., Шварц Дж. Т., Линейные операторы, пер. с англ., ч. 2-3, М., 1966-74; Келдыш М. В., Лидский В. Б., Вопросы спектральной теории несамосопряженных операторов, в кн.: Тр. 4-го Всесоюзного математического съезда, т. 1, Л., 1963, с. 101-20.
III
Спектра́льный ана́лиз
функции, обобщение гармонического анализа, тоже самое, что и Спектральное разложение функции.
Спектральный анализ
СТРАНИЦА ЗНАЧЕНИЙ В ПРОЕКТЕ ВИКИМЕДИА
Спектральный анализ (физич., химич.); Спектроскопический анализ
Спектральный анализ — совокупность методов качественного и количественного определения состава объекта, основанная на изучении спектров взаимодействия материи с излучением, включая спектры электромагнитного излучения, акустических волн, распределения по массам и энергиям элементарных частиц и др.
СПЕКТРАЛЬНЫЙ АНАЛИЗ
СТРАНИЦА ЗНАЧЕНИЙ В ПРОЕКТЕ ВИКИМЕДИА
Спектральный анализ (физич., химич.); Спектроскопический анализ
физический метод качественного и количественного определения состава вещества, проводимый по его спектрам оптическим. Различают атомный и молекулярный спектральный анализ, эмиссионный (по спектрам испускания) и абсорбционный (по спектрам поглощения). В качественном спектральном анализе полученный спектр интерпретируют с помощью таблиц и атласов спектров элементов и индивидуальных соединений; в количественном спектральном анализе определяют содержание исследуемого вещества по относительной или абсолютной интенсивностям линий или полос в спектрах. Применяется в промышленности, сельском хозяйстве, геологии и др.
Рентгеновский структурный анализ
методы исследования структуры вещества по распределению в пространстве и интенсивностям рассеянного на анализируемом объекте рентгеновского излучения. Р. с. а. наряду с нейтронографией (См. Нейтронография) и электронографией (См. Электронография) является дифракционным структурным методом; в его основе лежит взаимодействие рентгеновского излучения с электронами вещества, в результате которого возникает Дифракция рентгеновских лучей. Дифракционная картина зависит от длины волны используемых рентгеновских лучей (См. Рентгеновские лучи) и строения объекта. Для исследования атомной структуры применяют излучение с длиной волны Рентгеновский структурный анализ1 Å, т. е. порядка размеров атомов. Методами Р. с. а. изучают металлы, сплавы, минералы, неорганические и органические соединения, полимеры, аморфные материалы, жидкости и газы, молекулы белков, нуклеиновых кислот и т.д. Наиболее успешно Р. с. а. применяют для установления атомной структуры кристаллических тел. Это обусловлено тем, что Кристаллы обладают строгой периодичностью строения и представляют собой созданную самой природой дифракционную решётку для рентгеновских лучей.
Историческая справка. Дифракция рентгеновских лучей на кристаллах была открыта в 1912 немецкими физиками М. Лауэ, В. Фридрихом и П. Книппингом. Направив узкий пучок рентгеновских лучей на неподвижный кристалл, они зарегистрировали на помещенной за кристаллом фотопластинке дифракционную картину, которая состояла из большого числа закономерно расположенных пятен. Каждое пятно - след дифракционного луча, рассеянного кристаллом. Рентгенограмма, полученная таким методом, носит название лауэграммы (См. Лауэграмма) (рис. 1).
Разработанная Лауэ теория дифракции рентгеновских лучей на кристаллах позволила связать длину волны λ излучения, параметры элементарной ячейки кристалла а, b, с (см. Кристаллическая решётка), углы падающего (α0, β0, γ0) и дифракционного (α, β, γ) лучей соотношениями:
a (cosα- cosα0) = hλ,
b (cosβ - cosβ0) = kλ, (1)
c (cosγ - cosγ0) =lλ,
где h, k, I - целые числа (Миллеровские индексы). Для возникновения дифракционного луча необходимо выполнение приведённых условий Лауэ [уравнений (1)], которые требуют, чтобы в параллельных лучах разность хода между лучами, рассеянными атомами, отвечающими соседним узлам решётки, были равны целому числу длин волн.
В 1913 У. Л. Брэгг и одновременно с ним Г. В. Вульф предложили более наглядную трактовку возникновения дифракционных лучей в кристалле. Они показали, что любой из дифракционных лучей можно рассматривать как отражение падающего луча от одной из систем кристаллографических плоскостей (дифракционное отражение, см. Брэгга - Вульфа условие). В том же году У. Г. и У. Л. Брэгги впервые исследовали атомные структуры простейших кристаллов с помощью рентгеновских дифракционных методов. В 1916 П. Дебай и немецкий физик П. Шеррер предложили использовать дифракцию рентгеновских лучей для исследования структуры поликристаллических материалов. В 1938 французский кристаллограф А. Гинье разработал метод рентгеновского малоуглового рассеяния для исследования формы и размеров неоднородностей в веществе.
Применимость Р. с. а. к исследованию широкого класса веществ, производственная необходимость этих исследований стимулировали развитие методов расшифровки структур. В 1934 американский физик А. Патерсон предложил исследовать строение веществ с помощью функции межатомных векторов (функции Патерсона). Американские учёные Д. Харкер, Дж. Каспер (1948), У. Захариасен, Д. Сейр и английский учёный В. Кокрен (1952) заложили основы так называемых прямых методов определения кристаллических структур. Большой вклад в развитие патерсоновских и прямых методов Р. с. а. внесли Н. В. Белов, Г. С. Жданов, А. И. Китайгородский, Б. К. Вайнштейн, М. Порай-Кошиц (СССР), Л. Полинг, П. Эвальд, М. Бюргер, Дж. Карле, Г. Хауптман (США), М. Вульфсон (Великобритания) и др. Работы по исследованию пространственной структуры белка, начатые в Англии Дж. Берналом (30-е гг.) и успешно продолженные Дж. Кендрю, М. Перуцем, Д. Кроуфут-Ходжкин и др., сыграли исключительно важную роль в становлении молекулярной биологии (См. Молекулярная биология). В 1953 Дж. Уотсон и Ф. Крик предложили модель молекулы дезоксирибонуклеиновой кислоты (ДНК), которая хорошо согласовалась с результатами рентгенографических исследований ДНК, полученными М. Уилкинсом.
В 50-х гг. начали бурно развиваться методы Р. с. а. с использованием ЭВМ в технике эксперимента и при обработке рентгеновской дифракционной информации.
Экспериментальные методы Р. с. а. Для создания условий дифракции и регистрации излучения служат рентгеновские камеры (См. Рентгеновская камера) и рентгеновские дифрактометры (См. Рентгеновский дифрактометр). Рассеянное рентгеновское излучение в них фиксируется на фотоплёнке или измеряется детекторами ядерных излучений (См. Детекторы ядерных излучений). В зависимости от состояния исследуемого образца и его свойств, а также от характера и объёма информации, которую необходимо получить, применяют различные методы Р. с. а. Монокристаллы, отбираемые для исследования атомной структуры, должны иметь размеры Рентгеновский структурный анализ 0,1 мм и по возможности обладать совершенной структурой. Исследованием дефектов в сравнительно крупных почти совершенных кристаллах занимается Рентгеновская топография, которую иногда относят к Р. с. а.
Метод Лауэ - простейший метод получения рентгенограмм от монокристаллов. Кристалл в эксперименте Лауэ неподвижен, а используемое рентгеновское излучение имеет непрерывный спектр. Расположение дифракционных пятен на лауэграммах (рис. 1) зависит от симметрии кристалла (См. Симметрия кристаллов) и его ориентации относительно падающего луча. Метод Лауэ позволяет установить принадлежность исследуемого кристалла к одной и 11 лауэвских групп симметрии и ориентировать его (т. е. определять направление кристаллографических осей) с точностью до нескольких угловых минут. По характеру пятен на лауэграммах и особенно появлению Астеризма можно выявить внутренние напряжения и некоторые др. дефекты кристаллической структуры. Методом Лауэ проверяют качество монокристаллов при выборе образца для его более полного структурного исследования.
Методы качания и вращения образца используют для определения периодов повторяемости (постоянной решётки) вдоль кристаллографического направления в монокристалле. Они позволяют, в частности, установить параметры а, b, с элементарной ячейки кристалла. В этом методе используют монохроматическое рентгеновское излучение, образец приводится в колебательное или вращательное движение вокруг оси, совпадающей с кристаллографическим направлением, вдоль которого и исследуют период повторяемости. Пятна на рентгенограммах качания и вращения, полученных в цилиндрических кассетах, располагаются на семействе параллельных линий. Расстояния между этими линиями, длина волны излучения и диаметр кассеты рентгеновской камеры позволяют вычислить искомый период повторяемости в кристалле. Условия Лауэ для дифракционных лучей в этом методе выполняются за счёт изменения углов, входящих в соотношения (1) при качании или вращении образца.
Рентгенгониометрические методы. Для полного исследования структуры монокристалла методами Р. с. а. необходимо не только установить положение, но и измерить интенсивности как можно большего числа дифракционных отражений, которые могут быть получены от кристалла при данной длине волны излучения и всех возможных ориентациях образца. Для этого дифракционную картину регистрируют на фотоплёнке в рентгеновском гониометре (См. Рентгеновский гониометр) и измеряют с помощью Микрофотометра степень почернения каждого пятна на рентгенограмме. В рентгеновском дифрактометре (См. Рентгеновский дифрактометр) можно непосредственно измерять интенсивность дифракционных отражений с помощью пропорциональных, сцинтилляционных и других счётчиков рентгеновских квантов. Чтобы иметь полный набор отражений, в рентгеновских гониометрах получают серию рентгенограмм. На каждой из них фиксируются дифракционные отражения, на миллеровские индексы которых накладывают определённые ограничения (например, на разных рентгенограммах регистрируются отражения типа hk0, hk1 и т.д.). Наиболее часто производят рентгеногониометрический эксперимент по методам Вайсенберга. Бюргера (рис. 2) и де Ионга - Боумена. Такую же информацию можно получить и с помощью рентгенограмм качания.
Для установления атомной структуры средней сложности (Рентгеновский структурный анализ 50-100 атомов в элементарной ячейке) необходимо измерить интенсивности нескольких сотен и даже тысяч дифракционных отражений. Эту весьма трудоёмкую и кропотливую работу выполняют автоматические микроденситометры и дифрактометры, управляемые ЭВМ, иногда в течение нескольких недель и даже месяцев (например, при анализе структур белков, когда число отражений возрастает до сотен тысяч). Применением в дифрактометре нескольких счётчиков, которые могут параллельно регистрировать отражения, время эксперимента удаётся значительно сократить. Дифрактометрические измерения превосходят фоторегистрацию по чувствительности и точности.
Метод исследования поликристаллов (Дебая - Шеррера метод). Металлы, сплавы, кристаллические порошки состоят из множества мелких монокристаллов данного вещества. Для их исследования используют монохроматическое излучение. Рентгенограмма (дебаеграмма) поликристаллов представляет собой несколько концентрических колец, в каждое из которых сливаются отражения от определённой системы плоскостей различно ориентированных монокристаллов. Дебаеграммы различных веществ имеют индивидуальный характер и широко используются для идентификации соединений (в том числе и в смесях). Р.с.а. поликристаллов позволяет определять фазовый состав образцов, устанавливать размеры и преимущественную ориентацию (текстурирование) зёрен в веществе, осуществлять контроль за напряжениями в образце и решать другие технические задачи.
Исследование аморфных материалов и частично упорядоченных объектов. Чёткую рентгенограмму с острыми дифракционными максимумами можно получить только при полной трёхмерной периодичности образца. Чем ниже степень упорядоченности атомного строения материала, тем более размытый, диффузный характер имеет рассеянное им рентгеновское излучение. Диаметр диффузного кольца на рентгенограмме аморфного вещества может служить для грубой оценки средних межатомных расстояний в нём. С ростом степени упорядоченности (см. Дальний порядок и ближний порядок) в строении объектов дифракционная картина усложняется и, следовательно, содержит больше структурной информации.
Метод малоуглового рассеяния позволяет изучать пространственные неоднородности вещества, размеры которых превышают межатомные расстояния, т.е. составляют от 5-10 Å до Рентгеновский структурный анализ 10 000 Å. Рассеянное рентгеновское излучение в этом случае концентрируется вблизи первичного пучка - в области малых углов рассеяния. Малоугловое рассеяние применяют для исследования пористых и мелкодисперсных материалов, сплавов и сложных биологических объектов: вирусов, клеточных мембран, хромосом. Для изолированных молекул белка и нуклеиновых кислот метод позволяет определить их форму, размеры, молекулярную массу; в вирусах - характер взаимной укладки составляющих их компонент: белка, нуклеиновых кислот, липидов; в синтетических полимерах - упаковку полимерных цепей; в порошках и сорбентах - распределение частиц и пор по размерам; в сплавах - возникновение и размеры фаз; в текстурах (в частности, в жидких кристаллах) - форму упаковки частиц (молекул) в различного рода надмолекулярные структуры. Рентгеновский малоугловой метод применяется и в промышленности при контроле процессов изготовления катализаторов, высокодисперсных углей и т.д. В зависимости от строения объекта измерения производят для углов рассеяния от долей минуты до нескольких градусов.
Определение атомной структуры по данным дифракции рентгеновских лучей. Расшифровка атомной структуры кристалла включает: установление размеров и формы его элементарной ячейки; определение принадлежности кристалла к одной из 230 федоровских (открытых Е. С. Федоровым (См. Фёдоров)) групп симметрии кристаллов (См. Симметрия кристаллов); получение координат базисных атомов структуры. Первую и частично вторую задачи можно решить методами Лауэ и качания или вращения кристалла. Окончательно установить группу симметрии и координаты базисных атомов сложных структур возможно только с помощью сложного анализа и трудоёмкой математической обработки значений интенсивностей всех дифракционных отражений от данного кристалла. Конечная цель такой обработки состоит в вычислении по экспериментальным данным значений электронной плотности ρ(х, у, z) в любой точке ячейки кристалла с координатами x, у, z. Периодичность строения кристалла позволяет записать электронную плотность в нём через Фурье ряд:
где V - объём элементарной ячейки, Fhkl - коэффициенты Фурье, которые в Р. с. а. называются структурными амплитудами, i = 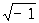 . Каждая структурная амплитуда характеризуется тремя целыми числами hkl и связана с тем дифракционным отражением, которое определяется условиями (1). Назначение суммирования (2) - математически собрать дифракционные рентгеновские отражения, чтобы получить изображение атомной структуры. Производить таким образом синтез изображения в Р. с. а. приходится из-за отсутствия в природе линз для рентгеновского излучения (в оптике видимого света для этого служит собирающая линза).
. Каждая структурная амплитуда характеризуется тремя целыми числами hkl и связана с тем дифракционным отражением, которое определяется условиями (1). Назначение суммирования (2) - математически собрать дифракционные рентгеновские отражения, чтобы получить изображение атомной структуры. Производить таким образом синтез изображения в Р. с. а. приходится из-за отсутствия в природе линз для рентгеновского излучения (в оптике видимого света для этого служит собирающая линза).
Дифракционное отражение - волновой процесс. Он характеризуется амплитудой, равной ∣Fhkl∣, и фазой αhkl (сдвигом фазы отражённой волны по отношению к падающей), через которую выражается структурная амплитуда: Fhkl =∣Fhkl ∣(cosαhkl + isinαhkl). Дифракционный эксперимент позволяет измерять только интенсивности отражений, пропорциональные ∣Fhkl∣2, но не их фазы. Определение фаз составляет основную проблему расшифровки структуры кристалла. Определение фаз структурных амплитуд в принципиальном отношении одинаково как для кристаллов, состоящих из атомов, так и для кристаллов, состоящих из молекул. Определив координаты атомов в молекулярном кристаллическом веществе, можно выделить составляющие его молекулы и установить их размер и форму.
Легко решается задача, обратная структурной расшифровке: вычисление по известной атомной структуре структурных амплитуд, а по ним - интенсивностей дифракционных отражений. Метод проб и ошибок, исторически первый метод расшифровки структур, состоит в сопоставлении экспериментально полученных ∣Fhkl∣эксп, с вычисленными на основе пробной модели значениями ∣Fhkl∣выч. В зависимости от величины фактора расходимости
пробная модель принимается или отвергается. В 30-х гг. были разработаны для кристаллических структур более формальные методы, но для некристаллических объектов метод проб и ошибок по-прежнему является практически единственным средством интерпретации дифракционной картины.
Принципиально новый путь к расшифровке атомных структур монокристаллов открыло применение т. н. функций Патерсона (функций межатомных векторов). Для построения функции Патерсона некоторой структуры, состоящей из N атомов, перенесём её параллельно самой себе так, чтобы в фиксированное начало координат попал сначала первый атом. Векторы от начала координат до всех атомов структуры (включая вектор нулевой длины до первого атома) укажут положение N максимумов функции межатомных векторов, совокупность которых называется изображением структуры в атоме 1. Добавим к ним ещё N максимумов, положение которых укажет N векторов от второго атома, помещенного при параллельном переносе структуры в то же начало координат. Проделав эту процедуру со всеми N атомами (рис. 3), мы получим N2 векторов. Функция, описывающая их положение, и есть функция Патерсона.
Для функции Патерсона Р (u, υ, ω) (u, υ, ω - координаты точек в пространстве межатомных векторов) можно получить выражение:
из которого следует, что она определяется модулями структурных амплитуд, не зависит от их фаз и, следовательно, может быть вычислена непосредственно по данным дифракционного эксперимента. Трудность интерпретации функции Р (u, υ, ω) состоит в необходимости нахождения координат N атомов из N2 её максимумов, многие из которых сливаются из-за перекрытий, возникающих при построении функции межатомных векторов. Наиболее прост для расшифровки Р (u, υ, ω) случай, когда в структуре содержится один тяжёлый атом и несколько лёгких. Изображение такой структуры в тяжёлом атоме будет значительно отличаться от др. её изображений. Среди различных методик, позволяющих определить модель исследуемой структуры по функции Патерсона, наиболее эффективными оказались так называемые суперпозиционные методы, которые позволили формализовать её анализ и выполнять его на ЭВМ.
Методы функции Патерсона сталкиваются с серьёзными трудностями при исследовании структур кристаллов, состоящих из одинаковых пли близких по атомному номеру атомов. В этом случае более эффективными оказались Так называемые прямые методы определения фаз структурных амплитуд. Учитывая тот факт, что значение электронной плотности в кристалле всегда положительно (или равно нулю), можно получить большое число неравенств, которым подчиняются коэффициенты Фурье (структурные амплитуды) функции ρ(x, у, z). Методами неравенств можно сравнительно просто анализировать структуры, содержащие до 20-40 атомов в элементарной ячейке кристалла. Для более сложных структур применяются методы, основанные на вероятностном подходе к проблеме: структурные амплитуды и их фазы рассматриваются как случайные величины; из физических представлений выводятся функции распределения этих случайных величин, которые дают возможность оценить с учётом экспериментальных значений модулей структурных амплитуд наиболее вероятные значения фаз. Эти методы также реализованы на ЭВМ и позволяют расшифровать структуры, содержащие 100-200 и более атомов в элементарной ячейке кристалла.
Итак, если фазы структурных амплитуд установлены, то по (2) может быть вычислено распределение электронной плотности в кристалле, максимумы этого распределения соответствуют положению атомов в структуре (рис. 4). Заключительное уточнение координат атомов проводится на ЭВМ Наименьших квадратов методом и в зависимости от качества эксперимента и сложности структуры позволяет получить их с точностью до тысячных долей Å (с помощью современного дифракционного эксперимента можно вычислять также количественные характеристики тепловых колебаний атомов в кристалле с учётом анизотропии этих колебаний). Р. с. а. даёт возможность установить и более тонкие характеристики атомных структур, например распределение валентных электронов в кристалле. Однако эта сложная задача решена пока только для простейших структур. Весьма перспективно для этой цели сочетание нейтронографических и рентгенографических исследований: нейтронографические данные о координатах ядер атомов сопоставляют с распределением в пространстве электронного облака, полученным с помощью Р. с. а. Для решения многих физических и химических задач совместно используют рентгеноструктурные исследования и резонансные методы.
Вершина достижений Р. с. а. - расшифровка трёхмерной структуры белков, нуклеиновых кислот и других макромолекул. Белки в естественных условиях, как правило, кристаллов не образуют. Чтобы добиться регулярного расположения белковых молекул, белки кристаллизуют и затем исследуют их структуру. Фазы структурных амплитуд белковых кристаллов можно определить только в результате совместных усилий рентгенографов и биохимиков. Для решения этой проблемы необходимо получить и исследовать кристаллы самого белка, а также его производных с включением тяжёлых атомов, причём координаты атомов во всех этих структурах должны совпадать.
О многочисленных применениях методов Р. с. а. для исследования различных нарушений структуры твёрдых тел под влиянием всевозможных воздействий см. в ст. Рентгенография материалов.
Лит.: Белов Н. В., Структурная кристаллография, М., 1951; Жданов Г. С., Основы рентгеноструктурного анализа, М. - Л., 1940; Джеймс Р., Оптические принципы дифракции рентгеновских лучей, пер. с англ., М., 1950; Бокий Г. Б., Порай-Кошиц М. А., Рентгеноструктурный анализ, М., 1964; Порай-Кошиц М. А., Практический курс рентгеноструктурного анализа, М., 1960: Китайгородский А. И., Теория структурного анализа, М., 1957; Липеон Г., Кокрен В., Определение структуры кристаллов, пер. с англ., М., 1961; Вайнштейн Б. К., Структурная электронография, М., 1956; Бэкон Дж., Дифракция нейтронов, пер. с англ., М., 1957; Бюргер М., Структура кристаллов и векторное пространство, пер. с англ., М., 1961; Гинье А., Рентгенография кристаллов, пер. с франц., М., 1961; Woolfson М. М., An introduction to X-ray crystallography, Camb., 1970: Ramachandran G. N., Srinivasan R., Fourier methode in crystallography, N. Y., 1970; Crystallographic computing, ed. F. R. Ahmed, Cph., 1970; Stout G. H., Jensen L. H., X-ray structure determination, N. Y. - L., [1968].
В. И. Симонов.
Рис. 1. Лауэграмма монокристалла NaCI. Каждое пятно представляет собой след рентгеновского дифракционного отражения. Диффузные радиальные пятна в центре вызваны рассеянием рентгеновских лучей на тепловых колебаниях кристаллической решётки.

Рис. 2. Рентгенограмма кристалла миоглобина.
Рис. 3. Схема построения функции Патерсона для структуры, состоящей из 3 атомов.
Рис. 9. а. Проекция на плоскость ab функции межатомных векторов минерала баотита [BA4Ti4 (Ti, Nb)4 [Si4O12] O16Cl]. Линии проведены через одинаковые интервалы значений функции межатомных векторов (линии равного уровня). б. Проекция электронной плотности баотита на плоскость ab, полученная расшифровкой функции межатомных векторов (a). Максимумы электронной плотности (сгущения линий равного уровня) отвечают положениям атомов в структуре. в. Изображение модели атомной структуры баотита. Каждый атом Si расположен внутри тетраэдра, образованного четырьмя атомами O; атомы Ti и Nb - в октаэдрах, составленных атомами O. Тетраэдры SiO4 и октаэдры Ti(Nb)O6 в структуре баотита соединены, как показано на рисунке. Часть элементарной ячейки кристалла, соответствующая рис. а и б, выделена штриховой линией. Точечные линии на рис. а и б определяют нулевые уровни значений соответствующих функций.
Рентгеноструктурный анализ
то же, что Рентгеновский структурный анализ.
Википедия
Рентгеноспектральный анализ
Рентгеноспектра́льный ана́лиз — инструментальный метод элементного анализа, основанный на изучении спектра рентгеновских лучей, прошедших сквозь образец или испущенных им.
