взаимное притяжение атомов, приводящее к образованию молекул и кристаллов. Принято говорить, что в молекуле или в кристалле между соседними атомами существуют Х. с. Валентность атома (о чём подробнее сказано ниже) показывает число связей, образуемых данным атомом с соседними атомами [см. также
Валентность]. Э.
Франкленд в 1852 предложил концепцию, согласно которой каждый элемент образует соединения, связываясь с определённым числом эквивалентов др. элементов, при этом один эквивалент соответствует количеству, требуемому одной валентностью. Ф. А.
Кекуле и А. В. Г.
Кольбе в 1857 в соответствии с представлениями валентности выдвинули положение, что углерод обычно имеет валентность 4, образует 4 связи с др. атомами. А. С.
Купер в 1858 указал, что атомы углерода, связываясь между собой, могут образовывать цепочки. В его записи химические формулы имели очень большое сходство с современными, связи изображались чёрточками, соответствующими валентным связям между атомами. Термин "химическое строение" впервые ввёл А. М.
Бутлеров в 1861. Он подчёркивал, сколь существенно выражать строение единой формулой, показывающей, как в молекуле соединения каждый атом связан с др. атомами. Согласно Бутлерову, все свойства соединения предопределяются его молекулярным строением; он высказал уверенность, что точную структурную формулу можно установить по результатам изучения путей синтеза данного соединения. Следующий шаг, заключавшийся в приписывании молекулам пространственной трёхмерной структуры, был сделан в 1874 Я. Х.
Вант-Гоффом и Ж. А. Ле Белем (См.
Ле Бель).
В 19 в. валентная связь изображалась чёрточкой между символами двух химических элементов. Природа этой связи была совершенно неизвестна. После открытия электрона делались многочисленные попытки развить электронную теорию Х. с. Наиболее успешными были работы Г. Н.
Льюиса, который в 1916 предложил рассматривать образование Х. с., называемой теперь ковалентной связью, как результат того, что пара электронов становится общей для двух атомов. Разработка квантовой механики (1925) и использование многих экспериментальных методов (молекулярной спектроскопии, рентгенографии кристаллов, газовой электронографии, методов изучения магнитных свойств) для определения длин связей (межатомных расстояний), углов между связями, числа неспаренных электронов и других структурных параметров молекул и кристаллов привели к более глубокому пониманию природы Х. с.
Электронная структура атомов. Электронам в атоме приписываются различные орбитали, которые характеризуются главным квантовым числом
n, орбитальным квантовым числом
l и магнитным квантовым числом
ml (см.
Квантовые числа,
Квантовая химия). Имеется одна наиболее устойчивая орбиталь с
n = 1, образующая
К-оболочку.
L-Оболочка с
n = 2 включает одну орбиталь с
l = 0 и
ml = 0 и три с
l = 1 и
ml = -1, 0 и +1. Их называют 1
s-орбиталь, 2
s-орбиталь и три 2
р-орбитали.
М-Оболочка состоит из 3
s-орбитали, трёх 3
р-орбиталей и пяти 3
d-орбиталей. Электрон имеет спин со спиновым квантовым числом
s =
1/
2, который может ориентироваться относительно определённого направления двумя различными путями - с компонентами, даваемыми магнитным спиновым квантовым числом
ms, равным +
1/
2 или -
1/
2. В атоме не может быть двух электронов с одинаковыми значениями всех квантовых чисел. Следовательно, 1
s-орбиталь, образующая
К-оболочку, может быть занята только одним электроном с положительным или отрицательным спином или же двумя электронами (электронной парой), одним - с положительным спином, другим - с отрицательным.
Заполнение определённых оболочек и подоболочек приводит к особой устойчивости атомов, наблюдающейся у атомов инертных газов. В этих устойчивых структурах электронная конфигурация заполненной оболочки гелия 1
s2, неона 2
s2 2
p6, аргона 3
s2 3
p6, криптона 3
d10 4
s2 4
p6, ксенона 4
d10 5
s2 5
p6, радона 4
f14 5
d10 6
s2 6
p6, эка-радона 5
f14 6
d10 7
s2 7
p6. [О заполнении электронных оболочек см. также
Атом,
Периодическая система элементов.]
Ковалентная связь. В 1927 датский физик О. Бурро выполнил квантовомеханический расчёт молекулярного нона водорода и показал, что единственный электрон в этом ионе На занимает орбиталь, называемую молекулярной орбиталью, которая простирается вокруг обоих протонов. Теоретический расчёт энергии связи этого молекулярного иона, т. е. разности между суммарной энергией отдельного атома и протона и энергией иона в его основном состоянии, привёл к значению 255 кдж․моль-1, прекрасно согласующемуся с экспериментом. Вскоре было отмечено, что электронную структуру молекулярного иона водорода можно рассмотреть, используя волновую функцию основного состояния атома водорода. По мере сближения атома водорода и протона появляется возможность выхода электрона из области, окружающей одно ядро, в область, окружающую второе ядро, причём в каждом случае электрон занимает 1s-орбиталь. Молекулярная орбиталь, образованная как сумма этих двух 1s-орбиталей, является хорошим приближением к молекулярной орбитали, полученной Бурро путём решения волнового уравнения Шрёдингера. Если образовать волновую функцию как разность двух 1s-орбиталей, то это, как было показано, отвечает не притяжению, а отталкиванию. Первая волновая функция является симметричной линейной комбинацией двух 1s-функций и отвечает устойчивому состоянию, образованию одноэлектронной ковалентной связи, тогда как вторая функция, являющаяся антисимметричной линейной комбинацией тех же 1s-функций, отвечает неустойчивому состоянию. Иногда говорят, что образование одноэлектронной ковалентной связи в молекуле водорода соответствует резонансу данного электрона между двумя атомными орбиталями или между двумя атомами водорода.
В том же году (1927) было выполнено два квантовомеханических расчёта Х. с. в молекуле водорода. Американский физик Э. У. Кондон использовал метод молекулярных орбиталей, приписав молекуле водорода структуру, в которой за основу была принята орбиталь H
2+, рассчитанная Бурро, причём к этой орбитали были отнесены оба электрона с противоположными спинами. Немецкие физики В.
Гейтлер и Ф. Лондон отнесли один электрон, с положительным спином, к 1
s-орбитали одного атома водорода, а второй, с отрицательным спином, к 1
s-орбитали др. атома водорода. Волновая функция для данной молекулы была суммой этой функции и функции, в которой два электрона менялись местами - электрон с положительным спином относился ко второму атому, а с отрицательным - к первому атому. Оба расчёта, как Кондона, так и Гейтлера и Лондона, привели к выводу об устойчивости молекулы водорода с энергией связи, превышающей приблизительно в 1,7 раза энергию связи в молекулярном ионе водорода. Связь между двумя атомами водорода в молекуле водорода - прототип связи с поделенной электронной парой по Льюису, обычно называют ковалентной связью.
На основании формальных результатов квантовомеханического рассмотрения Х. с. можно сделать следующий простой вывод: атомы могут образовывать ковалентную связь (осуществляемую парой электронов) за счёт каждой стабильной орбитали, занятой первоначально одним электроном; при этом образуется связь такого типа, как описанная выше для молекулы водорода, а её стабильность может быть связана с тем же самым явлением резонанса. Иными словами, для образования ковалентной связи необходимо наличие двух электронов с противоположными спинами и по одной стабильной орбитали у каждого из двух связываемых атомов.
Атом водорода с единственной стабильной орбиталью (1s) может образовывать лишь одну ковалентную связь. Атом углерода и другие атомы второго периода (бор, азот, кислород) могут образовывать не более четырёх ковалентных связей с использованием четырёх орбиталей L-оболочки. Квантовомеханическое рассмотрение приводит также к выводу, что каждая дополнительная связь, образующаяся в молекуле, в общем случае ведёт к дальнейшей стабилизации молекулы, а следовательно, наиболее устойчивы такие электронные структуры молекулы, в которых все стабильные орбитали атомов либо использованы для образования связей, либо заполнены неподелёнными парами электронов.
Метану CH4, например, приписывается следующая структура валентных связей:
Чёрточки означают поделенные электронные пары. Можно сказать, что поделенная электронная пара занимает 1s-орбиталь каждого атома водорода и одну из четырёх орбиталей L-оболочки атома углерода. Атомы водорода, т. о., комплектуют завершенную К-оболочку (как в атоме гелия), а атом углерода, который также имеет неподелённую пару 1s-электронов, комплектует завершенную L-оболочку (как в атоме неона).
Представление о гибридных орбиталях, формирующих связи, даёт решение проблемы, волновавшей химиков и физиков в ранний период квантовой теории. Четыре орбитали
L-оболочки делятся на два вида - 2
s-орбиталь и три 2
р-орбитали, а четыре связи атома углерода, как показывают химические свойства соединений углерода, оказываются одинаковыми. В действительности вместо 2
s-орбитали и трёх 2
р-орбиталей может образовываться набор эквивалентных
sp3-гибридных орбиталей, называется тетраэдрическими орбиталями; они направлены к вершинам правильного тетраэдра и обладают большей силой связи, чем
s-орбиталь или
р-орбиталь (Л.
Полинг,
1931).
Для молекулы воды H2O можно записать следующую валентную структуру:
Атом кислорода окружен двумя неподелёнными парами электронов и двумя поделенными парами. 2s-Орбиталь несколько более стабильна, нежели 2р-орбитали, так что неподелённые электронные пары прежде всего заполняют 2s-орбиталь. Если бы две связи в молекуле воды были образованы р-орбиталями атома кислорода, то угол между связями был бы равен 90°, поскольку при угле 90° друг относительно друга р-орбитали имеют максимальную силу связи. Расчёты показывают, что максимальная устойчивость достигается в том случае, когда орбитали, образующие связи в молекуле воды, в небольшой мере имеют также s-характер, соответственно валентный угол между связями несколько больший, чем 90°. Экспериментальное значение валентного угла в молекуле H2O 104,5°, а валентные углы в гидридах H2S, H2Se и H2Te равны 92, 91 и 90° соответственно.
Двойная ковалентная связь между атомами углерода имеется в этилене C2H4, а тройная связь - в ацетилене C2H2. Валентные структуры для этих молекул следующие:
В образовании двойной связи участвуют две поделенные электронные пары, а в образовании тройной связи - три пары. В каждой из этих структур атом углерода приобретает электронную конфигурацию неона, будучи окружен четырьмя поделенными парами электронов. Можно сказать, что атом углерода образует четыре одинарные (ординарные, простые) связи, направленные к вершинам тетраэдра. В двойной и тройной связях имеются две или три изогнутые связи. Интересно, что в этих случаях расстояния между атомами углерода равны соответственно 133 пм и 120 пм, что с точностью до 1 пм совпадает со значениями, соответствующими изогнутым связям при нормальной длине одинарной связи 154 пм в молекуле этана. Такое соответствие подтверждает правильность представления, что двойная и тройная связи могут быть описаны моделью изогнутых связей.
Энергия двойной углерод-углеродной связи на 73 кдж․моль-1 меньше, чем сумма энергий двух одинарных связей, энергия же тройной связи на 220 кдж․моль-1 меньше суммы энергий трёх одинарных связей. Эти различия в устойчивости могут быть связаны с напряжённостью изогнутых связей. Энергия напряжения благоприятствует превращению кратных связей в одинарные, и именно поэтому вещества с кратными связями легко присоединяют водород; такие вещества принято называть ненасыщенными, а соответствующие соединения, имеющие только одинарные связи, например этан, называются насыщенными.
Резонанс и структура бензола. Правила построения валентных структур на основании представлений о поделенных парах электронов и использования устойчивой орбитали каждого из двух атомов, между которыми образуется ковалентная связь, позволяют написать структурные формулы для очень большого числа веществ, однако для некоторых веществ одна валентная структура не даёт вполне адекватного представления о свойствах. Веществом именно такого рода является, например, озон O3. Спектроскопические исследования озона показали, что атомы в его молекуле расположены под углом 117° (угол между связями у центрального атома кислорода), а каждая из двух связей кислород - кислород имеет длину 128 пм. Есть все основания приписать молекуле озона следующую валентную структуру:
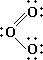
Эта структура представляется удовлетворительной, поскольку каждый из атомов кислорода окружен четырьмя парами электронов, причём некоторые пары поделенные, а некоторые неподелённые. Однако если приписать формальные заряды атомам, разделив поделенные пары электронов поровну между двумя атомами, то центральный атом будет иметь положительный заряд, а атом, связанный с ним одинарной связью, - отрицательный. Такую электронную структуру нельзя считать вполне удовлетворительной, поскольку межатомное расстояние, отвечающее двойной связи, должно быть приблизительно на 21 пм меньше, чем расстояние для одинарной связи, тогда как согласно наблюдениям эти расстояния равны. Такое расхождение можно объяснить, приняв и вторую валентную структуру для данной молекулы:
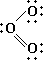
Приведённые структуры эквивалентны. При квантовомеханическом рассмотрении молекулы озона ей приписывается волновая функция, которая представляет собой сумму волновых функций для этих двух валентных структур. Установлено, что подобная волновая функция отвечает среднему значению длины связи, одному и тому же для обеих связей, и, кроме того, эта волновая функция соответствует большей стабильности, нежели каждая из волновых функций отдельных валентных структур. Такая дополнительная стабилизация описывается как энергия резонанса, соответствующая резонансу молекулы между двумя структурами. Отсюда следует, что озон нельзя удовлетворительно описать одной валентной структурой обычного типа, тогда как комбинация двух валентных структур приводит к удовлетворительному описанию молекулы в её основном состоянии.
Этот факт не противоречит основному принципу, выдвинутому в 1861 Бутлеровым, - каждое вещество имеет определённое молекулярное строение, которое обусловливает свойства данного вещества (см.
Химического строения теория,
Электронные теории в органической химии). Молекула озона в её основном состоянии имеет определённое единственное строение. Оно может быть представлено одной формулой:
Стрелки в этой формуле показывают, что двойная связь и одинарная связь могут меняться местами. Структура с двойной связью в одном положении и одинарной связью в другом не представляет какого-либо состояния молекулы озона, однако две резонирующие валентные структуры вместе взятые или структурная формула, в которой символически показано, что двойная и одинарная связи меняются местами, дают приемлемое представление о действительном единственном строении молекулы озона в основном состоянии.
Аналогичная ситуация наблюдается при рассмотрении молекулы бензола, строение которой казалось химикам загадочным до разработки (1928-33) теории резонанса (называемая также мезомерией). Кекуле указывал, что четырёхвалентность углерода в бензоле можно показать с помощью структурной формулы с чередующимися простыми и двойными связями. Однако таких структур может быть две:
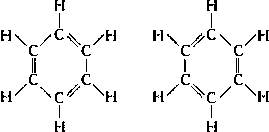
Были предприняты попытки обнаружить изомеры таких веществ, как о-дихлорбензол (атомы хлора присоединены к атомам углерода, связанным двойной связью в случае первого изомера и одинарной связью в случае второго). Однако обнаружить такие изомеры не удалось, и было признано, что все шесть углерод-углеродных связей в бензольном кольце эквивалентны друг другу. Детальное квантовомеханическое рассмотрение бензола показало. что его молекула имеет гексагональную симметрию и что все шесть углерод-углеродных связей эквивалентны. Этот факт позволяет сказать, что основное состояние молекулы бензола может быть представлено двумя структурами Кекуле, налагающимися одна на другую или резонирующими между собой. В соответствии с квантовомеханическими расчётами реальная молекула бензола должна быть приблизительно на 150 кдж․моль-1 устойчивее, нежели гипотетическая молекула, описываемая лишь одной структурой Кекуле. Эта дополнительная устойчивость обусловливает повышенную сопротивляемость бензола гидрогенизации по сравнению с обычными ненасыщенными соединениями.
Молекула бензола в её основном состоянии может быть представлена единственной формулой, такой, как:

. Кружок, проведённый внутри шестиугольника, означает, что данная структура описывает реальную молекулу, то есть отвечает большей устойчивости по сравнению со структурой Кекуле, и отражает эквивалентность всех шести углерод-углеродных связей. И всё же предпочтительнее бензол изображать двумя структурами Кекуле с оговоркой, что действительная структура молекулы соответствует резонансу между этими двумя структурами. Зная свойства, присущие одинарным связям и двойным связям, можно предсказать свойства, отвечающие структуре Кекуле и суперпозиции двух структур Кекуле. Длина одинарной углерод-углеродной связи 154
пм, а двойной связи - 133
пм. Для суперпозиции двух структур Кекуле ожидается среднее значение, более близкое, вследствие резонансной стабилизации, к значению для двойной связи. Наблюдаемое значение 140
пм согласуется с расчётным. Кроме того, если принять тетраэдрическую структуру каждого углеродного атома с деформированными (изогнутыми) двойными связями (общее ребро двух тетраэдров), можно предсказать, что молекула бензола должна быть плоской с атомами углерода в углах правильного шестиугольника и атомами водорода в углах большего правильного шестиугольника, лежащего в той же плоскости. Эти предсказания подтверждены опытными данными.
Ионная связь. Расплавленный хлорид натрия - хороший проводник электричества. Эту расплавленную соль можно считать состоящей из положительных ионов натрия Na+ и отрицательных ионов хлора Cl- в достаточно компактном состоянии, при котором в условиях термического равновесия каждый ион обладает возможностью медленно перемещаться. Под действием приложенного электрического поля ионы натрия передвигаются в направлении отрицательного электрода, а ионы хлора - в направлении положительного электрода, обусловливая проводимость электрического тока.
Ион натрия Na+ - это атом натрия, потерявший один электрон и приобретший устойчивую электронную конфигурацию неона, а ион хлора Cl- - атом хлора, присоединивший один электрон и приобретший устойчивую электронную конфигурацию аргона. Формула хлорида натрия NaCI определяется стабильностью этих ионов и условием электронейтральности данного вещества. Металлы первой группы периодической системы элементов Менделеева образуют однозарядные ионы и, как принято говорить, имеют ионную валентность +1; металлы второй группы образуют двухзарядные ионы и имеют ионную валентность +2, и т.д. Аналогично галогены, элементы седьмой группы, присоединяют электрон и образуют однозарядные отрицательные ионы, т. е. имеют ионную валентность -1; кислород и его аналоги могут присоединять два электрона с образованием двухзарядных отрицательных ионов со структурой инертных газов и обладают ионной валентностью -2, и т.д. Состав солей определяется ионными валентностями их катионов и анионов при соблюдении условия электронейтральности образующегося соединения.
Кулоновские силы, действующие между ионами, например Na+ и Cl-, приводят к тому, что каждый ион притягивает соседние ионы противоположного знака и создаёт из них окружение. В случае хлорида натрия это приводит к устойчивому упорядоченному расположению, отвечающему кристаллической структуре, при которой каждый ион имеет шесть ближайших соседей противоположного знака и двенадцать соседей того же знака, находящихся на расстоянии в 21/2 раза большем. Общая кулоновская энергия для такого расположения находится суммированием по парам ионов, и она равна -1,7476 e2/R для пары ионов Na+CI-, где R - расстояние между центрами ионов ближайших соседей, е - заряд иона. Следовательно, кристалл стабилизирован кулоновским притяжением, энергия такой системы на 75\% превышает энергию системы положительных и отрицательных зарядов, находящихся на тех же расстояниях R друг от друга. Кулоновская энергия кристалла NaCI большая - она составляет около 860 кдж․моль-1, с учётом сродства хлора к электрону затраты такой энергии более чем достаточно для сублимации металлического натрия, ионизации его атомов и диссоциации молекул хлора на атомы, а остальная энергия (410 кдж․моль-1) соответствует энергии образования хлорида натрия из элементов.
Силы притяжения ионов противоположного заряда называются силами ионной валентности. Можно сказать, что в кристалле хлорида натрия, в котором ион натрия имеет координационное число шесть (то есть он окружен шестью ближайшими соседями), общая ионная валентность иона натрия +1 разделяется между соседями, при этом каждую из шести связей между натрием и прилегающим хлором можно рассматривать как ионную связь силой 1/6. Отрицательный заряд иона хлора удовлетворяет шесть ионных связей, каждая силой 1/6, от шести соседних ионов натрия. Согласно правилу валентности, весьма существенному в неорганической химии, сумма ионных валентностей, направленных к каждому отрицательному иону, должна быть точно или приближённо равна ионной валентности данного отрицательного иона.
В ионных кристаллах связи в действительности не являются чисто ионными. Они носят частично ковалентный характер, о чём сказано в следующем разделе.
Электроотрицательность и частично ионный характер связей. В 20-х гг. 20 в.. когда были развиты концепции ионной валентности и ковалентности, но ещё не были известны основные принципы электронного строения атомов и молекул, велась широкая дискуссия о том, как описывать молекулу, подобную HCl - как имеющую ковалентную связь или как имеющую ионную связь. Структура H
+CI
- представлялась удовлетворительной, поскольку было известно о существовании соответствующих ионов, а ион хлора имеет устойчивую структуру аргона. Точно так же структура
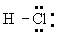
представлялась удовлетворительной, поскольку включала поделенную электронную пару, что создавало устойчивую конфигурацию гелия для водорода и устойчивую конфигурацию аргона для хлора. Хлористый водород в водном растворе диссоциирует на ионы водорода и хлора, а это позволяет предполагать, что ионное строение может быть присущим молекуле и в газовой фазе. Диэлектрическая проницаемость газа, однако, соответствует электрическому дипольному моменту, составляющему лишь 19\% величины, ожидаемой для ионной структуры при известном межатомном расстоянии 127
пм. Решение этой проблемы было найдено с помощью общей квантовомеханической теории молекулярного строения. Оно сводилось к тому, что действительное строение молекулы в основном состоянии может быть описано волновой функцией, представляющей собой сумму функций, отвечающих ионной структуре и ковалентной структуре. В случае молекулы HCl связь может быть описана как ионная со значительной долей ковалентности или, лучше сказать, как ковалентная связь с небольшой долей (19\%) ионности.
Рассматриваемая молекула в её основном состоянии имеет, конечно, единственное строение, которое может быть представлено единственной формулой Н-Cl. В случае ковалентной связи между одинаковыми атомами, как в Н-Н или Cl-Cl, связывающая электронная пара поделена поровну между двумя атомами. Идеальная ковалентная связь может быть определена как такая связь, в которой электронная пара поделена поровну между двумя атомами, даже если они не одинаковы. Если бы в HCl осуществлялась идеальная ковалентная связь, то можно было бы ожидать, что её энергия была бы средней между энергиями связей в H2 и Cl2. Действительно, для ряда одинарных связей между неодинаковыми атомами энергия связи равна средней энергии, отвечающей связям между одинаковыми атомами. Примером может служить HI с энергией связи 299 кдж․моль-1, которая всего лишь на 5 кдж․моль-1 больше среднего значения для H2 (436) и I2 (151). Электрический. дипольный момент молекулы HI также близок к нулю, а это указывает на то, что поделенная электронная пара почти в равной мере относится к обоим атомам. Связь в молекуле HI может быть описана как ковалентная с очень малой степенью ионности. Когда же связь имеет высокую степень ионности, энергия такой связи значительно превышает величину, отвечающую идеальной ковалентной связи; в случае HCl она на 92 кдж․моль-1 больше. Эта величина, представляющая собой энтальпию образования HCl из элементарных веществ, является энергией резонанса при 19\% ионности, т. е. энергией, соответствующей резонансу между ионной структурой и идеальной ковалентной структурой.
Было установлено, что одинарные связи между неодинаковыми атомами вообще несколько прочнее, чем средняя энергия соответствующих связей между одинаковыми атомами, и что этот выигрыш в энергии, энтальпии образования, в первом приближении пропорционален квадрату разности электроотрицательностей атомов. Значения электроотрицательности (х) могут быть приписаны элементам в соответствии с табл. (см.). Дополнительная энергия одинарной связи между неодинаковыми атомами приблизительно равна произведению 100 кдж․моль-1 на квадрат разности их электроотрицательностей. Несколько лучшее приближение достигается с учётом члена в четвёртой степени; тогда приближённое уравнение для энергии (Е) одинарной связи А-В (в кдж․моль-1) между различными атомами А и В будет иметь вид:
Для Н-Cl, например, это уравнение при E (H - Н) = 436, E (Cl - Cl) = 243 и xH - xCl = 0,9 даёт значение 417 кдж․моль-1, которое на 4\% меньше экспериментального значения 432 кдж․моль-1.
Наблюдаемые величины электрических дипольных моментов молекул показывают, что степень ионности связи А-В повышается с увеличением разности Δx = xA - xB и составляет приблизительно 22\% для Δх = 1,0, 63\% для Δх = 2,0 и 89\% для Δх = 3,0. Для HCl, например, наблюдаемое значение электрического дипольного момента составляет 19\% значения, соответствующего зарядам +2 и -2 при межъядерном расстоянии для молекулы 127 пм, что может быть сопоставлено со значением Δx = 0.9 для Н и Cl.
Полная шкала электроотрицательности*
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
| | | | | | | H | | | | | | | | | | |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| | | | | | | 2.1 | | | | | | | | | | |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| Li | Be | B | | | | | | | | | | | C | N | O | F |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| 1.0 | 1.5 | 2.0 | | | | | | | | | | | 2.5 | 3.0 | 3.5 | 4.0 |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| Na | Mg | Al | | | | | | | | | | | Si | P | S | Cl |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| 0.9 | 1.2 | 1.5 | | | | | | | | | | | 1.8 | 2.1 | 2.5 | 3.0 |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| 0.8 | 1.0 | 1.3 | 1.5 | 1.6 | 1.6 | 1.5 | 1.8 | 1.9 | 1.9 | 1.9 | 1.6 | 1.6 | 1.8 | 2.0 | 2.4 | 2.8 |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| 0.8 | 1.0 | 1.2 | 1.4 | 1.6 | 1.8 | 1.9 | 2.2 | 2.2 | 2.2 | 1.9 | 1.7 | 1.7 | 1.8 | 1.9 | 2.1 | 2.5 |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| Cs | Ba | La-Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| 0.7 | 0.9 | 1.0-1.2 | 1.3 | 1.5 | 1.7 | 1.9 | 2.2 | 2.2 | 2.2 | 2.4 | 1.9 | 1.8 | 1.9 | 1.9 | 2.0 | 2.2 |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| Fr | Ra | Ac | Th | Pa | U | Np-No | | | | | | | | | | |
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| 0.7 | 0.9 | 1.1 | 1.3 | 1.4 | 1.4 | 1.4-1.3 | | | | | | | | | | |
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
*По Полингу. Значения, приведённые в таблице, относятся к обычным окислительным состояниям элементов. Для некоторых элементов наблюдается изменение электроотрицательности с изменением окислительного числа; так, например, Fe (II)1,8, a Fe (III)1,9; Cu (I)1,9, а Cu (ll)2,0; Sn (ll)1,8, a Sn (IV)1,9.
Принцип электронейтральности. Принцип электронейтральности, впервые сформулированный И.
Ленгмюром (1920), гласит: устойчивые молекулы и кристаллы имеют такое электронное строение, при котором электрический заряд каждого атома близок к нулю, а по существу всегда лежит в пределах от -1 до +1. Так, например, степень ионности связи О-Н около 40\%, так что в молекуле воды H
2O результирующие заряды H
2+0,4 О
-0,6; в ионе гидроксония (H
3O)
+ результирующие заряды равны (H
3+0,4O
-0,2+)
+. Для молекулы закиси азота приемлемы следующие три структуры, поскольку они отвечают структуре неона для каждого атома:
(
А)
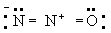
(
Б)
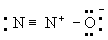
(
В)
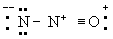
Однако третья структура не отвечает принципу электронейтральности, ибо формальный заряд -2 на концевом атоме азота не уравновешивается ионным характером связи N-N. Отсюда следует вывод, что нормальное состояние данной молекулы отвечает резонансу между структурами А и Б с очень небольшим вкладом или совсем без вклада структуры В; этот вывод подтверждается наблюдаемыми значениями длин связей и колебательных частот.
Окислительное число. После того как стали пользоваться представлениями об ионной валентности и ковалентности и начали подробно записывать электронное строение молекул, выявилась необходимость в простом способе указания окислительных состояний элементов в том или ином соединении. Для этой цели стали пользоваться понятием "окислительное число".
Окислительное число элемента в соединении выражает электрический заряд, приписываемый атомам данного элемента в соответствии с определёнными правилами. Эти правила простые, но не столь уж однозначные, и их применение требует химической интуиции: 1) окислительное число одноатомного иона в ионном соединении равно электрическому заряду данного иона. 2) Окислительное число атома в простом веществе равно нулю. 3) В ковалентном соединении известной структуры окислительное число каждого атома равно заряду, сосредоточенному на данном атоме при условии, что каждая поделенная пара электронов целиком приписывается более электроотрицательному из тех двух атомов, к которым она относится. Пара, относящаяся к атомам одного и того же элемента, обычно разделяется между ними. 4) Окислительное число элемента в соединении с неясной структурой может быть вычислено на основании разумного приписывания окислительных чисел другим элементам в данном соединении.
Например, в перекиси водорода H2O2 атомам водорода приписывается окислительное число +1; нейтральность молекулы требует тогда, чтобы кислород имел окислительное число -1. Следовательно, в H2O2 окислительное состояние кислорода - среднее между его состоянием в H2O (-2) и в O2 (0).
Слово "валентность" при его использовании в области неорганической химии обычно относится к состоянию окисления элемента, выражаемому окислительным числом, тогда как в органической химии оно обычно относится к ковалентности данного элемента.
Водородная связь. Структурным элементом, оказывающим значительное влияние на свойства многих веществ, является водородная связь. При определённых условиях атом водорода может быть связан довольно прочно с двумя др. атомами. Имея лишь одну стабильную орбиталь, атом водорода способен образовывать только одну ковалентную связь. Эта связь может, однако, резонировать между двумя положениями. Наибольшее значение имеют те водородные связи, которые образуются между двумя сильно электроотрицательными атомами, в особенности между атомами азота, кислорода и фтора. В некоторых соединениях, таких, как ион FHF-, атом водорода находится приблизительно посредине между двумя электроотрицательными атомами, образуя половину связи с каждым из них. Большинство же водородных связей несимметричны, одно межатомное расстояние больше другого на 50-80 пм, что соответствует отношению прочностей связи, равному приблизительно 10. Энергия более слабой связи обычно составляет около 20-40 кдж․моль-1, что и называется энергией водородной связи.
Водородные связи, образуемые молекулами воды, обусловливают удивительно высокие точки плавления льда и кипения воды, существование максимума плотности воды, расширение воды при замерзании. Многие особые свойства неорганических и органических молекул, например димеризация жирных кислот, объясняются образованием водородных связей. Водородная связь - особенно важная структурная особенность белков и нуклеиновых кислот.
Связи с участием d-орбиталей. В 1893 А.
Вернер развил новые представления в химии. Было известно, что многие соли металлов обладают способностью соединяться с др. солями, водой, аммиаком или др. молекулами. Хлорид калия и хлорид платины (IV) образуют, например, хорошо кристаллизующуюся соль 2KCI·PtCl
4, а иодид кобальта (III) присоединяет аммиак и образует CoI
3·6NH
3. Такого рода соединения, однако, не укладывались ни в одну теорию валентности, и их существование приписывалось действию слабых остаточных сил, второстепенных по сравнению с силами обычных Х. с. На основании изучения огромного числа таких соединений Вернер показал, что по составу и свойствам их можно систематизировать на базе нового допущения, согласно которому атом металла обладает способностью соединяться с определённым числом (обычно с четырьмя или шестью) др. атомов, ионов или молекул и координировать их вокруг себя в определённом геометрическом порядке. Вернер смог представить убедительные доказательства правильности своего предположения (оно подтверждалось главным образом фактом существования изомеров) о том, что большинство комплексов с координационным числом 6, таких, как гексахлороплатинат-ион [PtCl
6]
2- и гексаммино-кобальт (III)-ион [Со (NH
3)
6]
3+, имеют октаэдрическую конфигурацию, при которой шесть групп, окружающих центральный атом, располагаются вокруг него по вершинам правильного октаэдра. Он показал также, что ряд комплексов с координационным числом 4 имеет тетраэдрическую конфигурацию, например [Zn (NH
3)
4]
2+, тогда как другие - плоскую квадратную конфигурацию, характерную для комплексов Pd (II) и Pt (II), например для [PtCl
4]
2-. Общее признание теория Вернера получила в 1911, после его предсказания и экспериментального подтверждения существования оптической изомерии ряда октаэдрически координированных комплексов. В 1920 американские исследователи Р. У. Г. Уайкоф и Р. Г. Дикинсон рентгенографически определили структуры кристаллов K
2PtCI
6, K
2Pt (CN)
4 и др. координационных комплексов, окончательно подтвердив существование октаэдрических и плоскоквадратных конфигураций.
Теория этих комплексов была развита в 1931 Полингом. Он показал, что гибридизация s-орбитали и трёх р-орбиталей приводит к образованию четырёх тетраэдрических орбиталей, тогда как гибридизация этих четырёх орбиталей с двумя d-орбиталями приводит к набору из шести гибридных spd-орбиталей, направленных к вершинам правильного октаэдра, а с одной d-орбиталью образуются четыре гибридные sp2d-орбитали, направленные к вершинам квадрата. Число электронов в Pd (IV) и Pt (IV) таково, что две d-орбитали могут участвовать в образовании связи и, следовательно, образуются октаэдрические комплексы с координационным числом 6, тогда как Pd (ll) и Pt (ll) с двумя избыточными электронами имеют только одну доступную d-орбиталь и могут образовывать лишь квадратные плоские комплексы. Из такого рассмотрения вытекало, что ковалентные комплексы Ni (ll) должны иметь плоскую квадратную конфигурацию и быть диамагнитными, тогда как большинство соединений никеля парамагнитны. Эти предсказания сразу же были подтверждены результатами измерения магнитных свойств и определения кристаллической структуры координационных соединений никеля.
Химические связи в металлах. Природа Х. с. в металлах и интерметаллических соединениях остаётся и в 1977 выясненной не полностью. Представляется, однако, правильным описывать металлы и интерметаллические соединения как катионы металла, связанные воедино валентными электронами, обладающими значительной свободой движения в данном металле. Число электронов одного атома, участвующих в связывании металлического кристалла как целого, можно назвать "металлической валентностью" данного атома.
Металлическая валентность щелочных металлов 1, а щёлочноземельных 2. Значения для переходных металлов не вполне надёжны, однако, судя по прочности, твёрдости и точкам плавления, значения эти возрастают от 3 для Sc приблизительно до 6 для Cr и последующих элементов, а затем понижаются для Cu и Zn. Магнитные свойства лантаноидов свидетельствуют о том, что металлическая валентность их равна 3 (исключение составляют Eu и Yb, для которых она равна 2); парамагнитная восприимчивость Eu и Yb такая же, как и у их двухвалентных солей, тогда как для остальных лантаноидов она такая же, как у их трёхвалентных солей.
Координационное число атома в металле больше числа связывающих электронов. Связи в металлах могут быть описаны как ковалентные связи, резонирующие между некоторым большим числом межатомных положений. Так, например, алюминий имеет кубическую структуру с плотнейшей упаковкой, в которой каждый атом окружен двенадцатью соседями. Валентность алюминия равна 3 и, следовательно, связь с каждым из соседних атомов может быть описана как связь кратности 1/3.
Для того чтобы валентные связи могли резонировать между различными положениями, многие или большинство атомов должны иметь соответствующие орбитали связи, обычно не занятые электроном. Такие орбитали можно назвать "металлическими орбиталями". Характерной особенностью металлов является то, что большинство атомов в них обладают такой орбиталью. Олово, например, с четырьмя электронами на внешних s- и р-орбиталях может распределить эти четыре электрона между четырьмя sp3-орбиталями и образовать т. о. четыре ковалентные связи. Но тогда оно не будет иметь дополнительной орбитали и, следовательно, образующаяся структура не должна быть металлической. Модификация олова, называется серым оловом, действительно имеет структуру алмаза, в которой каждый атом связан с четырьмя тетраэдрически расположенными соседями и которая не является металлической. Длина связи здесь такая же, как длина одинарной связи. В белом олове, металлической модификации олова, каждый атом имеет шесть соседей с длиной связи, отвечающей валентности около 2,5 для атома олова. Если 2 из 4 внешних электронов атома олова образуют неподелённую пару, занимая 5s-орбиталь, то оставшиеся два электрона могут занять две из трех р-орбиталей и участвовать в образовании связи. При этом одна р-орбиталь остаётся свободной и может служить металлической орбиталью. По данным наблюдений, длина связи в белом олове отвечает металлической валентности 2,5, а не 2, что указывает на наличие резонанса (до 25\%) с четырёхвалентной структурой олова.
Если доступны d-орбитали, то могут образовываться гибридные spd-орбитали, которые ещё лучше подходят для образования связи, поскольку имеют большую концентрацию в направлении данной связи. В тех случаях, когда лучшие из возможных sp-орбиталей образуют между собой тетраэдрический угол 109°28', лучшие spd-орбитали образуют углы 73 и 133°.
Ковалентность переходных металлов. Переходные металлы с пятью d-орбиталями, одной s-орбиталью и тремя р-орбиталями во внешней оболочке могут образовывать 9 гибридных spd-орбиталей (под углами около 73 и 133° одна по отношению к другой) и, следовательно, могут образовывать 9 ковалентных связей в том случае, если данный атом имеет 9 электронов во внешней оболочке. Примером может служить Os4O4(CO)12. Структуру этого вещества можно описать как имеющую четыре атома осмия в четырёх противоположных вершинах куба и четыре атома кислорода в др. четырёх вершинах. Каждый атом кислорода передаёт электрон атому осмия. У этого атома кислорода, т. о., остаётся пять валентных электронов, и он может образовывать три ковалентные связи, а атом осмия имеет девять валентных электронов и может образовывать девять ковалентных связей. Каждый атом осмия образует три связи с прилегающими атомами кислорода и двойную связь с атомом углерода каждой из трех прилегающих карбонильных групп, достигая, т. о., своей максимальной валентности 9. Для большинства карбонилов переходных металлов химические формулы отвечают использованию всех 9 внешних spd-орбиталей для образования связей или неподелённых электронных пар. Например, атом никеля имеет 10 внешних электронов. В Ni (CO)4 8 из них используются для образования двойных связей с 4 карбонильными группами. На образование этих 4 двойных связей идут 8 из 9 spd-орбиталей, а оставшуюся одну занимает неподелённая пара. В Fe (CO)3 атом железа приобретает электрон от одной карбонильной группы, с которой он образует одинарную связь Fe-C≡O: оставшиеся 8 орбиталей и электроны он использует на образование двойных связей с атомами углерода четырёх других карбонильных групп. В Cr (СО)6 атом Cr получает 3 электрона от трёх карбонильных групп, что даёт 9 валентных электронов. Он образует одинарные связи с этими тремя группами и двойные связи с другими тремя карбонильными группами. Частично ионный характер хромуглеродных и углерод-кислородных связей, устанавливаемый по разности электроотрицательностей данных элементов, достаточен для передачи большей части избыточного отрицательного заряда электронов от хрома к кислороду так, что атомы остаются почти нейтральными, удовлетворяя принципу электронейтральности.
Четверные связи. Атомы углерода могут образовывать тройные связи, но не могут образовать четверных связей, поскольку четвёртая связь углерода направлена в сторону, противоположную направлению трёх остальных связей. Переходные металлы, однако, могут образовывать связи такой кратности благодаря тому, что четыре spd-орбитали под углом 73° друг к другу (около 133° для двух пар) направлены по одну сторону от атома. Первые данные о существовании таких связей были получены сов. химиками В. Г. Кузнецовым и П. А. Казьминым в 1963, когда они сообщили, что рентгеноструктурное изучение соединения рения показало присутствие группы Re2 с расстоянием Re-Re 222 пм, причём вокруг каждого атома рения располагалось четыре атома хлора на расстоянии 243 пм. Наблюдавшееся межатомное расстояние Re-Re приблизительно на 46 пм меньше, чем значение для одинарной связи. Очевидно, что в этом случае существует четверная связь, на что указывал в 1964 американский химик Ф. А. Коттон, который установил наличие аналогичных межатомных расстояний во многих др. кристаллах, а это подтверждает существование связей Cr≡Cr, Re≡Re, Tc≡Tc и Mo≡Mo.
Лит.: Полинг Л., Общая химия, пер. с англ., М., 1974; Паулинг Л. (Полинг), Природа химической связи, пер. с англ., М. - Л., 1947; Pauling L., The nature of the chemical bond and the structure of rnolecules and crystals..., 3 ed., lthaca (N. Y.), 1960.
Лайнус Полинг (США).
chromium-2D-skeletal.png?width=200 "Рис. 9. Дибензолхром")





 и длина одноэлектронной химической связи (d)")
![Рис. 14.[[Фазовая диаграмма]] водорода](https://commons.wikimedia.org/wiki/Special:FilePath/Phase diagram of hydrogen-ru.svg?width=200 "Рис. 14.[[Фазовая диаграмма]] водорода")







